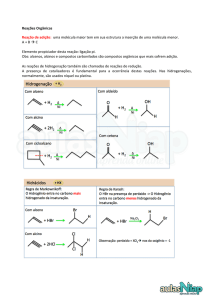
UNIVERSIDADE FEDERAL DO PARÁ
INSTITUTO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS FARMACÊUTICAS
RECRISTALIZAÇÃO POR EFEITO ANTI-SOLVENTE DE
UMA NOVA MOLÉCULA COM POTENCIAL EFEITO
CARDIOTÔNICO UTILIZANDO O GLICOFUROL COMO
SOLVENTE
ANTONIO TAYLON AGUIAR GOMES
BELÉM-PA
2014
UNIVERSIDADE FEDERAL DO PARÁ
INSTITUTO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS FARMACÊUTICAS
RECRISTALIZAÇÃO POR EFEITO ANTI-SOLVENTE DE
UMA NOVA MOLÉCULA COM POTENCIAL EFEITO
CARDIOTÔNICO UTILIZANDO GLICOFUROL COMO
SOLVENTE
Autor: Antonio Taylon Aguiar Gomes
Orientadora: Profª. Drª. Roseane Maria Ribeiro Costa
Co-orientadora: Profª. Drª. Maria Inês Ré
Dissertação apresentada ao Programa de Pós-Graduação em
Ciências Farmacêuticas, área de concentração: Fármacos e
Medicamentos, do Instituto de Ciências da Saúde da
Universidade Federal do Pará como requisito para a obtenção
do título de Mestre em Ciências Farmacêuticas.
BELÉM-PA
2014
Dados Internacionais de Catalogação-na-Publicação (CIP)
Biblioteca do Instituto de Ciências da Saúde – UFPA
Gomes, Antonio Taylon Aguiar
Recristalização por efeito anti-solvente de uma nova molécula com potencial efeito
cardiotônico utilizando o glicofurol como solvente / Antonio Taylon Aguiar Gomes ; orientadora,
Roseane Maria Ribeiro Costa ; co-orientadora, Maria Inês Ré. — 2014
Dissertação (Mestrado) - Programa de Pós- Graduação em Ciências Farmacêuticas –
Universidade Federal do Pará, Instituto de Ciências da Saúde (ICS), Belém, 2014.
1. Química farmacêutica. 2. Cristalização. 3. Excipientes farmacêuticos. 4. Dissolução.
5. Tamanho da partícula. I. Título.
CDD: 22..ed. : 615.19
FOLHA DE APROVAÇÃO
Antonio Taylon Aguiar Gomes
Recristalização por efeito anti-solvente de uma nova molécula com
potencial efeito cardiotônico utilizando glicofurol como solvente
Dissertação apresentada ao Programa de PósGraduação em Ciências Farmacêuticas do Instituto de
Ciências da Saúde da Universidade Federal do Pará
como requisito para a obtenção do título de Mestre em
Ciências Farmacêuticas.
Área de concentração: Fármacos e Medicamentos
Aprovado em: 30/09/2014
Banca Examinadora
Profª. Drª. Roseane Maria Ribeiro Costa
Instituição: UFPA/PPGCF
Assinatura:
Profª. Drª. Jacqueline Resende de Azevedo
Instituição: Ecole des Mines d’Albi-Carmaux, France
Assinatura:
Profª. Drª. Natália de Farias Silva
Instituição: UFPA/PPGCF
Assinatura:
“Quem vive de orgulho
morre de saudade”
Da Silva, W. O.
Resumo
A baixa solubilidade em água de muitos fármacos representa um obstáculo para
uma adequada biodisponibilidade oral. A redução das partículas de fármacos, entre
outras técnicas, tem sido descrita como uma estratégia para contornar esse
problema. A redução do tamanho de partícula pode conduzir a um significativo
aumento na cinética de dissolução do fármaco, promovendo um aumento
substancial na biodisponibilidade. Diante disso, o objetivo do trabalho é a
recristalização de uma nova molécula com potencial efeito cardiotônico (identificada
como molécula ‘A’), pelo método de cristalização por efeito anti-solvente, afim de
reduzir o tamanho de partícula e aumentar a sua cinética de dissolução. Para evitar
o uso de solventes orgânicos como os clorados, uma das metas do trabalho é de
demonstrar a viabilidade do uso de glicofurol como solvente para a molécula A. A
metodologia utilizada envolveu a determinação da solubilidade da molécula A em
glicofurol e em mistura glicofurol e água usada como anti-solvente. Ensaios de
recristalização foram realizados em misturas de glicofurol e água e o produto
recristalizado foi analisado por espectroscopia na região do infravermelho, difração
de raios-x, microscopia eletrônica de varredura e dissolução in vitro. Os resultados
obtidos mostraram que a molécula recristalizada apresenta a mesma estrutura
cristalina da molécula inicial e menor tamanho de partículas. No entanto, evidenciouse o problema de aglomeração das partículas que limitou a cinética de dissolução.
Palavras-chaves: Recristalização por efeito anti-solvente, Glicofurol, Redução de
tamanho de partículas, Dissolução.
Abstract
The poor solubility of many drugs in water is an obstacle to an adequate oral
bioavailability. The micronization of drugs, among other techniques, has been
described as a strategy to solve this problem. The particle size reduction can lead to
a significant increase in the dissolution kinetics of a drug, causing a substantial
increase in bioavailability. Thus, the aim of this work is the recrystallization of a new
molecule with potential cardiotonic effect (identified here as molecule 'A'), by the
method of crystallization by anti-solvent, in order to reduce the particle size and
increase its dissolution kinetics. To avoid the use of organic solvents such as
chlorinated, one of the targets of this study is to demonstrate the feasibility of using
glycofurol as a solvent for molecule A. The methodology involved the determination
of the solubility of molecule `A` in glycofurol and in the mixture of glycofurol and
water (used as anti-solvent). Recrystallization assays were performed in mixtures of
water and glycofurol and the recrystallized product was analyzed by infrared
spectroscopy, x-ray diffraction, scanning electron microscopy and in vitro dissolution.
The results showed that the recrystallized molecule has the same crystal structure of
the original molecule and smaller particle size. However, a problem of agglomeration
of smaller particles was observed which limited the dissolution kinetics.
Keywords:
Dissolution.
Anti-solvent
recrystallization,
Glycofurol,
Particle
size
reduction,
Lista de ilustrações
Figura 1 - Esquema de representação da cela unitária de cristais ............................ 18
Figura 2 - Processo de liberação do fármaco da forma farmacêutica sólida, adaptado
de Brown et al. (2004) ............................................................................................... 22
Figura 3 - Cálculo da eficácia de dissolução (COSTA, 2002). .................................. 25
Figura 4 - Esquema do processo de formação de partículas adaptado de Thorat e
Dalvi (2012) ............................................................................................................... 30
Figura 5 - Formula molecular do glicofurol. ............................................................... 34
Figura 6 - Esquema básico do funcionamento de um equipamento de CLAE: a reservatório de fase móvel; b - bomba de alta pressão; c - válvula de injeção da
amostra; d - fase estacionária (coluna); e - detector; f – registrador ......................... 40
Figura 7 - Espectro de infravermelho da molécula A ................................................. 48
Figura 8 - Forma estrutural da molécula A. ............................................................... 49
Figura 9 - TG e DTA da molécula A .......................................................................... 49
Figura 10 - Microscopia eletrônica de varredura da molécula A em diferentes
magnitudes A (121x), B(316x), C(450x), D(1270x). .................................................. 50
Figura 11 - Curva de calibração correlacionado a Área dos picos com a
concentração ............................................................................................................. 52
Figura 12 - Cromatogramas do Lauril sulfato de sódio 0,5% (verde), glicofurol a 0,1%
(vermelho) e molécula A a 2 µg/mL (azul)................................................................. 52
Figura 13 - Microscopia optica do recristalizado de molécula com água utilizando
glicofurol como solvente ............................................................................................ 57
Figura 14 - Gráfico da solubilidade de molécula A em diferentes misturas de
glicofurol/água. .......................................................................................................... 58
Figura 15 - Espectros de infravermelho da molécula A (preto) e de seus
recristalizados na proporção de 2-1 (vermelho), 1-1 (azul) e 1-2 (rosa) .................... 59
Figura 16 - Difração de raios-x da molécula A (preto) e de seus recristalizado na
proporção de 2-1 (vermelho), 1-1 (azul) e 1-2 (rosa) ................................................ 60
Figura 17 - Microscopia eletrônica de varredura do recristalizado na proporção 2-1
nas seguintes magnitudes: A(100x), B(2080x), C (12450x), D (13960x) .................. 61
Figura 18 - Microscopia eletrônica de varredura do recristalizado na proporção 1-1
nas seguintes magnitudes: A(80x), B(3360x), C (1300x), D (10170x) ...................... 61
Figura 19 - Microscopia eletrônica de varredura do recristalizado na proporção 1:2
nas seguintes magnitudes: A(196x), B(621x), C (3870x), D (10710x) ...................... 62
Figura 20 - Microscopia eletrônica de varredura da molécula A original (A) e
recristalizada na proporção de 1-1 (B) na magnitude de 1270x. ............................... 62
Figura 21 - Perfil de dissolução da molécula A (vermelho) e de seu recristalizado
(azul) usando como meio água ................................................................................. 64
Figura 22 - Perfil de dissolução da Molécula A (azul) e de seu recristalizado
(vermelho) usando como meio LSS 0,5%. ................................................................ 65
Figura 23 - Molécula A em LSS 0,5% no modelo Peppas F=k*t^n ............................ 67
Figura 24 - Recristalizado em água no modelo Peppas F=k*t^n ............................... 67
Figura 25 - Recristalizado em LSS 0,5% no modelo Peppas F=k*t^n ....................... 68
Lista de tabelas
Tabela 1 - Sistemas cristalinos e a relação de vetores e ângulos ............................. 18
Tabela 2 - Classificação da solubilidade (PORTUGAL, 2002) .................................. 20
Tabela 3 - Sistema de Classificação Biofarmacêutica ............................................... 29
Tabela 4 - Fármacos recristalizados por processo de cristalização por efeito antisolvente usando solventes orgânicos ........................................................................ 33
Tabela 5 - Preparo das soluções para a curva de calibração ................................... 41
Tabela 6 - Preparação das soluções em diferentes proporções de glicofurol/água .. 44
Tabela 7 - Proporção de solução saturada e água para a recristalização. ................ 46
Tabela 8 - Curvas de calibração preparadas em 3 dias diferentes. .......................... 51
Tabela 9 - Repetibilidade do padrão de concetração de 2 µg/mL ............................. 53
Tabela 10 - Resultado da precisão intermediaria ...................................................... 54
Tabela 11 - Dados da recuperação do método ......................................................... 54
Tabela 12 - Adição de molécula A em glicofurol. ...................................................... 55
Tabela 13 - Solubilidade da molécula A em misturas de glicofurol e água. .............. 56
Tabela 14 - Quantificação da molécula A em diferentes meios saturados ................ 56
Tabela 15 - Cálculo do rendimento da produção de cristais ..................................... 58
Tabela 16 - Perfil de dissolução da molécula A em água e em LSS 0,5% ................ 63
Tabela 17 - Cálculo da eficácia de dissolução .......................................................... 66
Tabela 18 - Cálculo dos r² de alguns modelos matemáticos ..................................... 66
Lista de abreviaturas e siglas
As
CLAE
Cs
Área Superficial do Fármaco
Cromatografia Líquida de Alta Eficiência
Solubilidade máxima no meio de dissolução
Ct
CV%
D
dC/dt
DL50
DP
DRX
DTA
ED%
f1
f2
FT-IR
H
IC
k
Kdd
Concentração do fármaco no tempo t
Coeficiente de variação
Coeficiente de difusão
Taxa de Dissolução
Dose Letal para 50% dos indivíduos
Desvio padrão
Difração de Raio-X
Análise térmica diferencial
Eficácia de dissolução
Fator de diferença
Fator de similaridade
Infravermelho com transformadas de Fourier
Espessura da camada de difusão
Inclinação da reta
Coeficiente de dissolução ou transferência de massa
Velocidade de desintegração da formulação
Kdi
LD
LQ
LSS
MEV
PBS
PEG
pH
R
Rdt
rpm
SCB
SM
T
TG
V
Velocidade de dissolução intrínseca do fármaco
Limite de detecção
Limite de quantificação
Lauril sulfato de sódio
Microscopia Eletrônica de Varredura
Fosfato de Sódio Monobásico
Polietileno glicol
Potencial hidrogeniônico
Recuperação
Rendimento
Rotação por minuto
Sistema de classificação biofarmacêutica
Solução mãe
Tempo
Termogravimetria
Volume do meio de dissolução
Lista de símbolos e unidades
°C
µg
µm
Au
Br
Cu
g
K
Kg
KV
mA
mg
mL
mm
mv/s
nm
Graus célsius
Micrograma
Micrometros
Ouro
Bromo
Cobre
Grama
Potássio
Quilograma
Quilovolts
Miliampere
Miligrama
Mililitros
Milímetros
Milivolts por segundo
Nanômetros
SUMÁRIO
1
INTRODUÇÃO .......................................................................................... 15
2
REFERENCIAL TEÓRICO........................................................................ 17
2.1
Propriedades físico-químicas do fármaco .................................................... 17
Estrutura cristalina ........................................................................ 17
Tamanho da partícula................................................................... 19
Solubilidade .................................................................................. 19
Dissolução .................................................................................... 21
Ensaios de dissolução .................................................................. 23
2.2
Técnicas para melhorar a biodisponibilidade ............................................... 29
Sistema de classificação biofarmacêutica. ................................... 29
Cristalização por efeito anti-solvente ............................................ 30
Escolha do solvente e anti-solvente ............................................. 32
Glicofurol ...................................................................................... 33
3
OBJETIVOS .............................................................................................. 36
3.1
Objetivo Geral: ............................................................................................. 36
3.2
Objetivos Específicos: .................................................................................. 36
4
MATERIAL E MÉTODOS.......................................................................... 37
4.1
Caracterização da molécula A ..................................................................... 37
Espectroscopia na região do infravermelho com transformadas de
Fourier (FT-IR) ................................................................................................... 37
Análise Térmica ............................................................................ 37
Microscopia eletrônica de varredura ............................................ 38
Difração de raios-X ....................................................................... 39
4.2
Estudo da solubilidade ................................................................................. 39
Validação de um método de quantificação da molécula A em
solução por cromatografia liquida de alta eficiência (CLAE) .............................. 39
Solubilidade aparente da molécula A em glicofurol e em misturas
de glicofurol/água ............................................................................................... 44
Solubilidade “real” da molécula A em glicofurol e em misturas de
glicofurol/água .................................................................................................... 45
4.3
Preparação de cristais através da preciptação por anti-solvente ................. 45
Ensaio preliminar de cristalização ................................................ 45
Preparação dos cristais ................................................................ 45
Determinação da melhor proporção água/glicofurol para a
recristalização da molécula A............................................................................. 46
4.4
Caracterização do produto recristalizado ..................................................... 47
Perfil de dissolução ...................................................................... 47
5
RESULTADOS E DISCUSSÃO ................................................................ 48
5.1
Caracterização da molécula A ..................................................................... 48
Espectrometria de infravermelho com transformadas de fourier
(FT-IR)
48
Análise térmica ............................................................................. 49
Microscopia eletrônica de varredura ............................................ 50
5.2
Estudo da solubilidade ................................................................................. 51
Validação do mÉtodo de quantificação da molÉcula A por CLAE 51
Solubilidade aparente ................................................................... 55
Solubilidade real da molécula A em misturas de glicofurol/água.. 56
5.3
Preparação de cristais através da cristalização da molécula A em mistura
glicofurol/água ....................................................................................................... 57
Ensaio de cristalização ................................................................. 57
Determinação da melhor proporção glicofurol/água para a
recristalização da molécula A............................................................................. 58
5.4
Caracterização da molécula A e de seus recristalizados ............................. 59
Infravermelho com Tranformada de Fourier ................................. 59
Difração de Raio-X ....................................................................... 60
Microscopia Eletrônica de Varredura ........................................... 60
Dissolução .................................................................................... 63
6
CONCLUSÃO ........................................................................................... 69
7
PERSPECTIVAS E SUGESTÃO DE CONTINUIDADE ............................ 70
8
REFERÊNCIAS ........................................................................................ 71
15
1
INTRODUÇÃO
A molécula A, um fármaco em desenvolvimento industrial, é um derivado Nacilidrazônico com um potente efeito cardiotônico. Este novo fármaco tem a
capacidade de contrair o músculo cardíaco e promover vasodilatação através de um
mecanismo diferente dos glicosídeos cardíacos. Isso torna esse fármaco
interessante, por que os glicosídeos apresentam muitos efeitos que podem causar
arritmia e possuem baixo índice terapêutico (AZEVEDO, 2014a; BARREIRO, 2002).
Esse
novo
fármaco,
assim
como
70%
dos
medicamentos
em
desenvolvimento, apresenta uma baixa solubilidade em meio aquoso. Essa
propriedade causa uma dissolução lenta que compromete a biodisponibilidade da
molécula (MOU et al., 2011). Para contornar este tipo de problema, diferentes rotas
tecnológicas têm sido desenvolvidas para alterar a solubilidade e/ou a cinética de
dissolução destes fármacos tais como complexação, alteração da composição do
solvente, modificações químicas ou físicas (KAWABATA et al., 2011).
Uma das modificações físicas para o aumento da cinética de dissolução é a
redução do tamanho das partículas do fármaco (JINNO et al., 2006). Essa
transformação pode ser obtida por processos do tipo “top-down” (fragmentação de
partículas maiores até o tamanho desejado) ou do tipo “bottom-up” (formação de
cristais a partir de moléculas associadas via cristalização ou precipitação).
Processos do tipo “bottom-up” consomem menos energia e permitem, através do
controle do processo de formação do sólido, melhor manipular a morfologia e a
dispersão de tamanho do mesmo. Daí a importância de explorar esta rota para a
redução do tamanho das partículas (VERMA; GOKHALE; BURGESS, 2009).
Dentre as técnicas do tipo “bottom-up” a recristalização por efeito antisolvente é uma técnica de fácil aplicação e apresenta bons resultados (THORAT;
DALVI, 2012). Ela baseia-se na adição de um solvente em que o fármaco é insolúvel
(geralmente água, no caso de fármacos com baixa solubilidade) a uma solução onde
o fármaco encontra-se dissolvido em um bom solvente para o mesmo.
A incorporação de um anti-solvente à solução contendo o fármaco dissolvido
em um solvente mais adequado reduz a solubilidade do fármaco na mistura
16
resultante solvente/anti-solvente. Ao ser atingida a saturação inicia-se a formação de
pequenos núcleos (nucleação) seguida do crescimento destes núcleos (cristais).
Uma vez formados, estes cristais podem mudar de tamanho por aglomeração ou
quebra. A formação dos cristais por cristalização por efeito anti-solvente requer a
identificação de um solvente para o fármaco em questão, assim como o
conhecimento de sua solubilidade neste solvente e a variação da solubilidade na
mistura de solvente com o anti-solvente. O controle das características do sólido
gerado depende do controle da velocidade de nucleação do sistema, da cinética de
crescimento e de aglomeração dos cristais formados (ROSSMANN et al., 2012).
Os principais desafios do ponto de vista tecnológico para o desenvolvimento
de um produto de interesse farmacêutico através de um processo de cristalização
por efeito anti-solvente são:
O controle da forma sólida formada (amorfa x cristalina);
O controle da cinética de nucleação para formação dos cristais;
O entendimento e o controle da cinética de crescimento e aglomeração
para controle do tamanho final destes cristais;
Controle da aglomeração dos cristais em suspensão e controle da
estabilidade física destes sistemas (no caso de nanosuspensões).
O solvente escolhido para a solubilização do molécula A foi o glicofurol que é
um solvente atóxico e cujo potencial em solubilizar outras moléculas
de baixa
solubilidade em água tem sido demonstrada (ALLHENN; LAMPRECHT, 2011;
AUBERT-POUËSSEL et al., 2004; BOONGIRD et al., 2011; BURY et al., 1984;
VIEHOF et al., 2013). Com base neste estado da arte, avaliou-se neste trabalho, a
capacidade do glicofurol em solubilizar a molécula A e de seu uso como solvente
para uma recristalização da molécula em presença de água como anti-solvente.
17
2
2.1
REFERENCIAL TEÓRICO
Propriedades físico-químicas do fármaco
Toda substância tem um conjunto de propriedades que as caracteriza. As
propriedades químicas são aquelas observadas quando a substância sofre uma
alteração e converte-se em uma ou mais substâncias. As propriedades físicas são
as propriedades que podem ser medidas sem alterar a identidade química da
substância. Como exemplo dessas propriedades tem-se a densidade, ponto de
fusão, solubilidade e a cor (MASTERTON; SLOWINSKI; STANITSKI, 1990).
Essas propriedades são importantes para o desenvolvimento de um
medicamento, pois elas podem afetar tanto a formulação do produto como o seu
comportamento no organismo (ATTWOOD; FLORENCE, 2003). O tamanho das
partículas, a existência de polimorfos e a higroscopicidade são exemplos de
características do fármaco que podem alterar a absorção do medicamento
(STORPIRTIS; MARCOLONGO, 2004). Diante disso, faz-se necessário realizar o
estudo de pré-formulação durante a concepção de um novo medicamento.
O estudo de pré-formulação consiste nessa etapa de pesquisa das
propriedades físico-químicas de um fármaco isolado ou composto e sua
compatibilidade com excipientes (ALVES-SILVA; SÁ-BARRETO, 2014; DURIG;
FASSIHI, 1991). As principais áreas a serem investigadas na pré-fomulação de um
fármaco sólido é a caracterização do sólido (como pesquisa de polimorfos), análise
da solubilidade e a análise da estabilidade (LACHMAN; LIEBERMAN; KANIG, 2001).
ESTRUTURA CRISTALINA
Um fármaco pode existir em diferentes formas cristalinas. Esta habilidade do
sólido é chamada polimorfismo (ARAUJO; JR, 2012). Além de diferentes polimorfos,
o fármaco pode apresentar-se como amorfo (não apresenta nenhuma estrutura
cristalina definida) ou como solvatos (quando apresenta solventes adsorvidos em
sua estrutura). Quando o solvente adsorvido é a água esse solvatos comumente são
chamados de hidratos (RAW et al., 2004). Diferentes polimorfos da mesma
18
substância podem apresentar diferentes características física e químicas e isso pode
alterar a tanto a estabilidade do fármaco como a sua biodisponibilidade (XIE et al.,
2008). Em geral, a forma amorfa do fármaco é mais solúvel facilitando a
biodisponibilidade, porém é menos estável que a forma cristalina dificultando a
armazenagem do fármaco (GUO; SHALAEV; SMITH, 2013). Dessa maneira o
conhecimento das formas cristalinas é uma ferramenta muito importante para o
estudo da pré-formulação.
Quando na forma de cristais as moléculas do fármaco se agrupam de maneira
organizada, onde a menor célula de organização é chamada de cela unitária (Figura
1) e é definida pelos vetores de translacionais a, b e c e os ângulos α,β e ɣ formando
sete sistemas cristalinos (Tabela 1): cúbico, tetragonal, ortorrômbico, romboédrico ou
trigonal, hexagonal, monoclínico e triclínico (ARAUJO; JR, 2012).
Figura 1 - Esquema de representação da cela unitária de cristais
Tabela 1 - Sistemas cristalinos e a relação de vetores e ângulos
Sistema
Vetores
Cubico
a=b=c
Tetragonal
a=b≠c
Ortorrômbico a≠b≠c
Romboédrico a=b=c
Hexagonal
a=b≠c
Monoclínico
a≠b≠c
Triclínico
a≠b≠c
Ângulos
α=β=ɣ=90°
α=β=ɣ=90°
α=β=ɣ=90°
α=β=ɣ≠90°
α=β=90° e ɣ=120°
α=ɣ=90°≠β
α≠β≠ɣ≠90°
19
As principais técnicas para o estudo do polimorfismo de um fármaco são a
difração
de
raios-X,
termogravimetria,
calorimetria
exploratória
diferencial,
espectroscopia Raman e na região do infravermelho e microscopia polarizada
(ZHANG et al., 2004).
TAMANHO DA PARTÍCULA
O
tamanho
da
partícula
influencia
inversamente
a
dissolução
do
medicamento, ou seja, quanto menor o tamanho da partícula maior será a
dissolução. Isso ocorre por que quanto menor o tamanho da partícula maior será a
superfície de contato do medicamento para interagir com o meio. Em certos casos a
redução de fármacos lipossolúveis pode diminuir a dissolução por formar agregados
(BRANDÃO, 2003).
A estabilidade do fármaco também é alterada quando se diminui o tamanho
das partículas pois ele fica mais reativo, podendo alterar a aparência do
medicamento e a uniformidade (MINÉ; MORAIS, 2013).
SOLUBILIDADE
A solubilidade é um parâmetro termodinâmico que representa a concentração
da solução de uma substância em equilíbrio com o soluto a uma dada temperatura.
Pode ser determinada através da adição de um excesso de fármaco ao meio, sob
agitação até que o equilíbrio seja atingido, seguido de filtração e quantificação do
fármaco dissolvido. A solubilidade pode ser afetada pela natureza do solvente, do
soluto e da temperatura. É o fator que mais afeta a velocidade de dissolução
(VIÇOSA et al., 2012).
As farmacopeias brasileira (BRASIL, 2010a), portuguesa (PORTUGAL, 2002)
e britânica (PHARMACOPOEIA, 2009) classificam a solubilidade das substâncias
em muito solúvel, facilmente solúvel, solúvel, ligeiramente solúvel, pouco solúvel,
muito pouco solúvel e praticamente insolúvel através do número de partes
(quantidade de mililitros) necessário para solubilizar 1g da substância. A Tabela 2
20
mostra a classificação da solubilidade segundo a farmacopeia portuguesa
(PORTUGAL, 2002).
Tabela 2 - Classificação da solubilidade (PORTUGAL, 2002)
Descrição
Quantidade de solvente em mililitros para 1
grama da substância
Muito solúvel
menos de
1
Facilmente solúvel
De
1 a 10
Solúvel
De
10 a 30
Ligeiramente solúvel
De
30 a 100
Pouco solúvel
De
100 a 1000
Muito pouco solúvel
De
1000 a 10000
mais de
10000
Praticamente insolúvel
Um dos primeiros fatores a serem estudados durante o desenvolvimento de
um novo fármaco é a solubilidade (MOTA et al., 2011). No organismo ela pode variar
em função de parâmetros fisiológicos, como o pH do trato gastrintestinal (SHARGEL;
YU, 1999). A solubilidade de um fármaco é um critério chave nos estudos de préformulação, ou seja, na etapa de investigação na qual se identificam as
propriedades físicas e químicas de fármacos e excipientes que podem influenciar a
formulação, a forma farmacêutica, o processo produtivo e as propriedades
farmacocinéticas do medicamento.
Há duas décadas atrás, os fármacos insolúveis em água eram rejeitados
devido à falta de técnicas para a sua formulação. Entretanto, cerca de 70% dos
novos fármacos são insolúveis em água (KAWABATA et al., 2011). Atualmente a
farmacotécnica obteve avanços importantes na preparação de medicamentos
possibilitando a formulação desses fármacos insolúveis, necessitando apenas que
esses estejam solubilizados em um solvente orgânico (LIU, 2008).
21
Para a solubilização de fármacos com baixa solubilidade em água existem
algumas tecnologias como: micronização, formação de nanocristal por moinho de
bola ou tecnologia de gás denso, solução sólida (incorporada em polímeros),
soluções lipídicas, emulsões e sistema de surfactante e co-solvente (POUTON,
2006).
DISSOLUÇÃO
Em sistemas biológicos, a dissolução de medicamentos pode ser definida
como o processo pelo qual um fármaco é liberado de sua forma farmacêutica,
tornando-se disponível para ser absorvido pelo organismo (MISRA et al., 2012). A
dissolução dependerá das propriedades do fármaco (ex: pKa, solubilidade, tamanho
das partículas) e das propriedades do trato gastrointestinal (ex: volume e pH dos
fluidos, taxa de fluxo, taxa de cisalhamento)(MUDIE; AMIDON; AMIDON, 2010).
Nas formas farmacêuticas sólidas, o medicamento tem que se solubilizar e
atravessar várias barreiras do organismo para chegar ao seu sítio de ação. Assim, o
estudo da dissolução do fármaco é de fundamental importância para se entender
como será seu comportamento no organismo (SIEPMANN; SIEPMANN, 2013).
Os processos relacionados à liberação do fármaco de formas farmacêuticas
sólidas estão relacionados na Figura 2, como proposto na literatura (BROWN et al.,
2004), onde Kdd representa a velocidade de desintegração da formulação e Kdi a
velocidade de dissolução intrínseca do fármaco.
22
Farmaco na
formulação
Kdi
Kdd
Farmaco em
particulas
Farmaco na
solução
Figura 2 - Processo de liberação do fármaco da forma farmacêutica sólida, adaptado de
Brown et al. (2004)
Constata-se que quando Kdd é maior que Kdi, a dissolução é controlada pela
dissolução intrínseca dos ativos, sendo suas propriedades físico-químicas
consideradas importantes. Já quando Kdd é menor que Kdi, a dissolução é controlada
pela desintegração, sendo esta influenciada pelas propriedades coesivas da
formulação (BROWN et al., 2004).
A absorção sistêmica geralmente é limitada para fármacos com baixa
solubilidade em água que se dissolvem a partir da forma farmacêutica, intacta ou
desintegrada, no trato gastrintestinal. Para estes fármacos, a etapa de dissolução
atua como um limitante que determina, consequentemente, a velocidade de
absorção e a quantidade absorvida, o que se traduz em valores incorretos de
absorção (WONG; KELLAWAY; MURDAN, 2006).
Um dos desafios mais relevantes na rotina de desenvolvimento de
formulações de liberação imediata contendo fármacos pouco solúveis concentra-se
no controle da velocidade de dissolução e liberação do ativo disponível.
23
ENSAIOS DE DISSOLUÇÃO
Os testes in vitro são de fundamental importância para se predizer o que
ocorrerá com o fármaco nos testes in vivo (KOVAČIČ et al., 2014). Cada fármaco e
cada uma das suas formas farmacêuticas apresentam um tipo de teste de
dissolução utilizando um determinado aparato e uma especifica quantidade de meio
(USP, 2006).
Um aparelho de dissolução apresenta três partes: as cubas (um cilindro
aberto com fundo hemisférico); as hastes de aço inoxidável (que, em geral, pode ser
no formato de uma cesta ou de uma pá); e um motor (promotor da rotação das
hastes). Para o teste utiliza-se um meio de dissolução e que é determinado de
acordo com o material a ser analisado, a temperatura deve ser de 37°C ± 0,5°C. A
velocidade de rotação das hastes, a quantidade de amostra de respeitar a condição
“sink” (até 20% da concentração de saturação) e o tempo da dissolução é especifico
para cada fármaco (BRASIL, 2010a) (MANADAS; PINA; VEIGA, 2002).
2.1.5.1 Teoria da dissolução de fármacos
Em 1897, Noyes e Whitney estabeleceram uma Equação (1), com base na
segunda lei de difusão de Fick, relacionando a velocidade de dissolução com a
solubilidade máxima do soluto (ou constante de saturação) e a concentração ao
tempo t (MANADAS; PINA; VEIGA, 2002):
(1)
Onde, dC/dt é a taxa de dissolução, K o coeficiente de dissolução ou de
transferência de massa, Cs a solubilidade máxima no meio de dissolução, Ct a
concentração do fármaco na solução no tempo t; e (Cs – Ct) o gradiente de
concentração.
Em 1904, Nernst e Brunner modificaram a equação de Noyes e Whitney
formando a Equação (2), tendo incluído como parâmetros influentes no processo o
24
coeficiente de difusão (D), a área superficial do fármaco (As), a espessura da
camada de difusão (h) e o volume do meio de dissolução (V) (MANADAS; PINA;
VEIGA, 2002):
(2)
A Equação 2 demonstra que a dissolução é influenciada pelas características
físico-químicas da substância ativa, pelo solvente e pela formulação, englobando a
natureza dos excipientes e o processo produtivo. Contudo, em relação à
disponibilidade do fármaco no organismo, particularmente do trato gastrintestinal,
fatores como a permeabilidade através da membrana do epitélio e o coeficiente de
partição também irão afetar a capacidade do fármaco em ser absorvido (SHARGEL;
YU, 1999).
Para aumentar a velocidade de dissolução a partir da equação de Nernst e
Brunner as principais rotas possíveis são o aumento de As pela redução do tamanho
de partículas do fármaco. Um aumento na área As aumenta a velocidade de
dissolução.
2.1.5.2 Modelos cinéticos
Existem 3 tipos de testes de dissolução: ensaio de dissolução de um único
ponto, ensaio de dissolução de 2 pontos e perfil de dissolução. O perfil de dissolução
é o teste mais completo pois fornece a porcentagem do fármaco dissolvido em
diferentes pontos da dissolução (CHORILLI; SOUZA, 2010).
Para a avaliação do perfil de dissolução podem ser utilizadas 3 tipos de
analises: método baseado na análise de variância, método modelo independente e
método modelo dependente (SERRA; STORPIRTIS, 2007).
Método análise de variância
O método de análise de variância utiliza os dados na sua forma nativa, não
necessitando a plotagem de gráficos. Após a aplicação do teste estatístico ANOVA
frequentemente faz-se um pós-teste para esclarecer quais grupos diferem, já que a
ANOVA não faz essa distinção. Os teste estatísticos mais comuns utilizados no pós-
25
testes são: Turkey, Bonferroni, Newman-Keuls e Dunnet (RODRIGUES; SILVA,
2005).
Modelos independentes
Eficácia de dissolução (ED%)
A eficácia de dissolução é um cálculo que mostra quanto do fármaco foi
solubilizado na solução através da área que ele forma no gráfico do perfil de
dissolução. Esse cálculo (Figura 3) não esclarece o comportamento da dissolução
do fármaco, mas evidência sua performance em comparação a outro perfil de
dissolução (COSTA, 2002).
Figura 3 - Cálculo da eficácia de dissolução (COSTA, 2002).
Fator de diferença (f1) e fator de similaridade (f2)
O fator de diferença e o fator de similaridade são usados para comparação
entre perfis de dissolução. Utilizando a equação do fator de diferença pode-se dizer
que os perfis de dissolução são iguais quando estão próximos a 0 e são aceitáveis
valores que variam de 0 a 15. Para o fator de similaridade afirma-se que os perfis de
dissolução são iguais quando próximo a 100 e são aceitáveis valores que variam de
50 a 100 (COSTA, 2002, 2001; COSTA; SOUSA LOBO, 2001). Para o cálculo do f1
utiliza-se a Equação (3) e para o cálculo do f2 a Equação (4).
26
(3)
(4)
Onde,
Rj= quantidade de molécula A dissolvida no tempo J
Tj= quantidade de recristalizado dissolvido no tempo j
N= número de recolhas
Modelos dependentes (mecanismo de liberação)
O comportamento cinético da dissolução dos fármacos pode ser determinado
através de equações matemáticas. A escolha do modelo cinético pode ser feita
utilizando a correlação de Pearson (r2), ou seja, o modelo que mais se adequa
aquela curva é aquele em que os pontos estão mais próximos aos da equação,
assim quanto mais próximo de 1 for o r2 maior será a adequação do perfil de
dissolução a aquele modelo cinético proposto (SCHESHOWITSCH; PEREIRA,
2007).
Primeira ordem
Este modelo descreve medicamentos que liberam fármacos de forma
proporcional a quantidade que está em seu interior, de modo que ao passar do
tempo a quantidade de liberação vai diminuindo. Fármacos hidrossolúveis em
matrizes porosas é um exemplo de medicamento que se comporta nesse modelo
(MANADAS; PINA; VEIGA, 2002).
(5)
27
A Equação 5 descreve modelo cinético de primeira ordem onde, Qt é a
quantidade de fármaco dissolvida no tempo t, Q 0 é a quantidade do fármaco inicial
na solução.
Higuchi
O modelo de Higuchi foi desenvolvido para descrever fármacos solúveis e
pouco solúveis incorporados em matrizes solidas e/ou semissólidas. Este modelo
tem sido muito utilizado para descrever fármacos de liberação controlada (COSTA,
2002; PAUL, 2011).
(6)
Equação 6, onde KH é a constante de Higuchi baseada na lei de Fick
(MANADAS; PINA; VEIGA, 2002).
Hixon e Crowell
A equação de Hixon e Crowell é baseada na premissa de que a superfície do
fármaco é proporcional a raiz cúbica do seu volume. A diminuição da superfície de
contato ocorre de forma proporcional a que o fármaco vai se solubilizando e assim
vai mantendo sua geometria até completa solubilização. Medicamentos que estão
ligados a esse modelo tem sua velocidade de liberação do fármaco limitada pela
velocidade de dissolução do fármaco (SINGHVI; SINGH, 2011).
(7)
Equação 7, onde Q0 é a quantidade de fármaco no medicamento, Q t é a
quantidade de fármaco presente no medicamento no tempo t e K hc é a constante de
Hixon e Crowell.
28
Peppas
Este modelo é utilizado para descrever liberação de medicamentos sob a
forma farmacêutica poliméricas, quando o mecanismo de liberação não é conhecido
ou existe mais de um mecanismo envolvido na liberação do fármaco (COSTA, 2002).
(8)
Equação 8, onde a é uma constante que leva em consideração as
características estruturais e geométricas da forma farmacêutica, n é o expoente de
liberação determinado pelo mecanismo de liberação (MANADAS; PINA; VEIGA,
2002).
Baker e Lonsdale
Com base na equação do modelo de Higuchi foi desenvolvida essa equação
que descreve fármacos de liberação controlada na forma de matriz esférica (COSTA;
SOUSA LOBO, 2001). A equação de Baker e Lonsdale (Equação 9) onde, Mt é a
quantidade de fármaco no medicamento no tempo t, M∞ é a quantidade de fármaco
no medicamento no tempo infinito, Dm é o coeficiente de dissolução, Cms é a
solubilidade do fármaco na matriz, r0 é o raio da esfera da matriz e C0 é a
concentração inicial do fármaco na matriz.
(9)
29
2.2
Técnicas para melhorar a biodisponibilidade
SISTEMA DE CLASSIFICAÇÃO BIOFARMACÊUTICA.
O sistema de classificação biofarmacêutica (SCB) introduzido por Amidon et
al. (1995) permite predizer a absorção dos fármacos segundo a sua solubilidade em
meio aquoso e permeabilidade biológica. Segundo os critérios adotados, um
fármaco pode ser classificado de acordo com a Tabela 3:
Tabela 3 - Sistema de Classificação Biofarmacêutica
Classe 1
Classe 2
Classe 3
Classe 4
Solubilidade
Alta
Baixa
Alta
Baixa
Permeabilidade
Alta
Alta
Baixa
Baixa
A classificação SCB é de extrema importância, uma vez que mais de 80% do
mercado farmacêutico mundial é representado por formulações administradas por
via oral, sendo ainda a exigência de uma biodisponibilidade adequada, pré-requisito
legal em quase todo o mundo, inclusive no Brasil (BRASIL, 2010b).
Os fármacos da classe 1 tem uma boa biodisponibilidade via oral. Os
fármacos da classe 3 e 4, devido à baixa permeabilidade, geralmente sofrem uma
modificação química em suas estruturas para melhorar a sua biodisponibilidade
(POUTON, 2006). Os da classe 2 são fármacos que precisam melhorar a sua
solubilidade. Nesse último caso, diferentes técnicas têm sido aplicadas para
contornar esse problema, dentre elas (KAWABATA et al., 2011):
Modificações nos cristais
Diminuição do tamanho da partícula
Amorfização
Complexação com ciclodextrina
Auto-emulsificação
Modificações no pH
30
A equação de Noyes-Whitney mostra que quanto maior a superfície de
contato maior será a razão de dissolução do fármaco. Assim, através da diminuição
do tamanho da partícula, pode-se aumentar a dissolução do fármaco por causa do
aumento da área de superfície de contato. Uma das técnicas para diminuir o
tamanho das partículas é a cristalização por anti-solvente (JINNO et al., 2006).
CRISTALIZAÇÃO POR EFEITO ANTI-SOLVENTE
A
cristalização
por
efeito
anti-solvente
baseia-se
na
mudança
da
supersaturação causada pela mistura da solução com um anti-solvente. Ela ocorre
através das etapas: mistura da solução com o anti-solvente, produção da
supersaturação, formação de núcleos cristalinos e crescimento a partir dos núcleos
formados e, em alguns casos, aglomeração (Figura 4).
Figura 4 - Esquema do processo de formação de partículas adaptado de Thorat e Dalvi
(2012)
A supersaturação é definida como a força matriz da cristalização que depende
da concentração de saturação (Cs) e da concentração do fármaco na solução (Ct)
Equação (10) (KAKRAN et al., 2012).
31
(10)
A nucleação compreende a etapa de aparecimento dos novos cristais na
solução, os quais conduzem a formação de germes ou núcleos que irão crescer.
Existem 2 tipos de nucleação: a primária e a secundária. A nucleação primária
ocorre sem a presença de um material cristalino e pode ser dividida em homogênea
(quando a nucleação ocorre de forma espontânea a partir da união das moléculas)
ou heterogênea (quando a nucleação é induzida por materiais estranhos como
poeira). A nucleação secundária ocorre com a presença de cristais na solução
(AALTONEN et al., 2009).
Logo que os núcleos são formados, eles começam a crescer através da
incorporação de moléculas de soluto na interface sólido-líquido. Ao contrário da
nucleação e crescimento, a aglomeração não ocorre em todos os processos de
cristalização. Ela depende das condições do sistema e do processo de cristalização.
Tendo em vista, que a formação de cristais se produz imediatamente graças à
rápida desolvatação da molécula, após a mistura da solução com o anti-solvente,
considera-se que o ponto chave na produção de partículas ultrafinas por
precipitação anti-solvente é criar condições que favoreçam a formação rápida das
partículas (nucleação) e pouco ou nenhum crescimento dos núcleos formados,
evitando problemas de aglomeração (THIERING; DEHGHANI; FOSTER, 2001).
De acordo com a literatura, inúmeros fatores podem influenciar as
propriedades dos cristais formados, dentre eles: modo de mistura, concentração da
solução, proporção solvente/anti-solvente, temperatura e velocidade de agitação. O
efeito da concentração do fármaco na obtenção de cristais foi descrito por Dong et
al. (2009). Nesse estudo, o tamanho de partícula foi reduzido com o aumento da
concentração do fármaco na solução. Zhang et al. (2009) também descreveram esse
comportamento quando a concentração foi modificada de 20 mg/mL para 60 mg/mL.
No entanto, quando a concentração foi aumentada para 80 mg/mL notou-se a
formação de aglomerados. Os resultados descritos estão relacionados com o grau
de supersaturação. O efeito da proporção solvente/anti-solvente foi descrito por
Zhang et al. (2006) e Zhao et al. (2007). Nos dois casos, o tamanho de partícula foi
32
reduzido com o aumento da proporção de anti-solvente na mistura. Isto foi explicado
pela diminuição da solubilidade à medida que a quantidade de anti-solvente na
mistura aumenta (aumento da supersaturação).
A cristalização por efeito anti-solvente pode proporcionar uma flexibilidade
maior para o controle de formas amorfas e cristalinas do fármaco e é uma técnica de
fácil implementação. Esta técnica já foi testada com sucesso para produção de
partículas ultrafinas de fármacos pouco solúveis em água como a budesonida
(RASENACK; HARTENHAUER; MÜLLER, 2003), danazol (ZHAO et al., 2007),
dipropionato de beclometasona (WANG; CHEN; LE, 2007), predinisolona (LI et al.,
2007), diclofenaco (LAI; SINICO; ENNAS, 2009), griseofulvina e fenofibrato (MENG;
CHEN; CHOWDHURY, 2009).
ESCOLHA DO SOLVENTE E ANTI-SOLVENTE
Apesar da cristalização por efeito anti-solvente ser investigada com bastante
interesse na área científica, ela apresenta algumas desvantagens que limitam o
interesse e a exploração industrial como o uso de solventes orgânicos voláteis
(acetona, DMSO, dentre outros) na solubilização dos fármacos, como mostra a
Tabela 4.
Como limitações para a utilização de solventes na precipitação por efeito antisolvente pode-se mencionar:
Adição, na maioria dos casos, de um solvente inflamável e poluente em
meio aquoso;
Necessidade de separação posterior de solventes por destilação de
alto custo;
Caso o solvente seja tóxico, há a preocupação com a remoção do
solvente no produto acabado, que após secagem, deve conter
solventes em níveis limites aceitáveis, pois muitos solventes orgânicos
são tóxicos e de uso limitado na área farmacêutica (DHIRENDRA et al.,
2009).
33
Tabela 4 - Fármacos recristalizados por processo de cristalização por efeito antisolvente usando solventes orgânicos
Fármaco
Fase orgânica
Fase aquosa
Estabilizante
Autor
Cefuroxima
axetil
Acetato de etila/
Cloridrato de
metileno/
Cloroformio/ Acido
fórmico/ éter
isopropilico/
acetona
Eter
isopropílico/
água
X
(ZHANG et al.,
2006)
Itraconazol
Tetrahidrofurano
Poly/ P407
X
(MATTEUCCI et al.,
2006)
Espirolactona
Bicalutamida
Itraconazol Odanacatib
Metanol/ etanol/
isopropanol/
acetona
Água
Etanol/ DMSO
Água
Tween 80/
SDS/
Pluronic F127/ álcool
polivinil/
HPMC
X
Tetrahidrofuran
Água
PS-b/ PEO
Água
SLS/ PVPK30/ Pluronic
F-127/ Tween
80/ HPMC
Ibuprofeno
Acetona
(DONG et al., 2009)
(LE et al., 2009)
(KUMAR; WANG;
RIEBE, 2009)
(VERMA;
GOKHALE;
BURGESS, 2009)
GLICOFUROL
O glicofurol a-[(Tetrahydro-2-furanyl)methyl]-o-hydroxy-poly(oxy-1,2-ethanediyl) é um solvente orgânico límpido, incolor, quase sem odor e produz uma
sensação de ardência na língua. É miscível em água e não é tóxico, onde apresenta
uma DL50=3,5 mL/Kg. Sua fórmula molecular é C9H18O4 e apresenta a formula
estrutural de acordo com a Figura 5 (ROWE; SHESKEY, 2006). A toxicidade do
glicofurol foi estudada por Boongird et al. (2011) após à injeção de glicofurol no
cérebro de ratos com a injeção de solução de tampão fosfato. Todos os ratos
sobreviveram e não houve uma significante perda de massa. Nenhuma anomalia
neurológica foi observada. Sendo assim, os autores concluíram que o glicofurol é um
solvente biocompatível.
34
Figura 5 - Formula molecular do glicofurol.
Tauboll et al. (1990) estudaram a solubilização da carbamazepina em
glicofurol para uso parenteral. Nos experimentos a carbamazepina apresentou uma
solubilidade de 100 mg/mL em glicofurol e de 150 mg/mL na mistura glicofurol:álcool
benzílico (1:1) à temperatura ambiente. As soluções permaneceram estáveis por
mais de 14 dias e em animais mostraram que carbamazepina em glicofurol
mantiveram a atividade farmacológica. O glicofurol também foi utilizado em
formulações injetáveis contendo fármacos insolúveis, tais como: diazepam e
fenitoína (IVATURI et al., 2009), genes (ELIAZ; SZOKA, 2002) e proteínas
(BAGGER; NIELSEN; BECHGAARD, 2001). Além de injetável esse solvente
também foi utilizado como promotor da penetração de fármacos em uso tópico,
solvente para soluções intranasal e retais (BOONGIRD et al., 2011)
A utilização de glicofurol como solvente também foi descrita na preparação de
microesferas. Aubert-Pouessel et al. (2004) utilizaram esse solvente na obtenção de
microesferas de lisozima através da emulsificação. As micropartículas tiveram
diâmetros em torno de 10 µm. Allhenn E Lamprecht (2011) descreveram a
preparação de microesferas de fármacos insolúveis como ibuprofeno, ritonavir e
lopinavir. A utilização do glicofurol como solvente justificou-se pelo fato da técnica de
preparação
de
microesferas
por
evaporação
de
solvente
frequentemente
apresentava resquícios do solvente utilizado, que geralmente era tóxico, ao contrário
do glicofurol que é um solvente atóxico. Os resultados demonstraram que a
utilização de glicofurol como solvente na preparação de microesferas desses
fármacos insolúveis é uma alternativa satisfatória.
Outro estudo descreveu o uso do glicofurol como co-solvente na preparação
de um gel de naproxeno (BARAKAT, 2010). O naproxeno é um anti-inflamatório com
35
baixa solubilidade em água, assim é inviável a preparação de um hidrogel para a
sua administração. Porém, utilizando o glicofurol para a solubilização do fármaco e o
carbopol para introduzir as características de gel, o autor conseguiu um produto com
boa permeabilidade, adesividade e espalhabilidade.
Viehof et al. (2013) prepararam nanopartículas de insulina utilizando como
solvente o glicofurol e polietileno glicol (PEG). Além de serem atóxicos esses
solventes não desnaturam a proteína como ocorre na preparação com acetona,
tetrahidrofurano ou etanol. O tipo de solvente não influenciou as propriedades das
partículas ou a estabilidade da insulina, mas modificou significativamente a liberação
in vivo em ratos. As nanopartículas preparadas com glicofurol apresentaram maior
biodisponibilidade após administração oral que as obtidas com PEG 300. Os autores
concluíram que os métodos apresentados para preparação de nanopartículas de
insulina são promissores para encapsulação de outros fármacos sensíveis.
Dessa forma, a utilização do glicofurol como solvente alternativo para
solubilização da molécula A é justificada pela sua baixa toxicidade e pela sua
capacidade de solubilizar fármacos insolúveis. Além disso, devido a sua
miscibilidade com agua, este solvente é adequado para uso em processos de
recristalização, onde a água é utilizada como anti-solvente.
36
3
3.1
OBJETIVOS
Objetivo Geral:
Recristalizar uma nova molécula com potencial efeito cardiotônico, denominada
molécula A, pela técnica de cristalização por efeito anti-solvente em presença de
glicofurol, afim de melhorar sua cinética de dissolução.
3.2
Objetivos Específicos:
Caracterizar físico-quimicamente a molécula A;
Desenvolver um método de quantificação da molécula A em solução por
cromatografia liquida de alta eficiência;
Determinar a solubilidade da molécula A em glicofurol e em misturas de
glicofurol/água;
Recristalizar a molécula A utilizando o glicofurol como solvente e água
como anti-solvente;
Caracterizar os cristais obtidos e comparar com os da molécula inicial.
37
4
4.1
MATERIAL E MÉTODOS
Caracterização da molécula A
ESPECTROSCOPIA
NA
REGIÃO
DO
INFRAVERMELHO
COM
TRANSFORMADAS DE FOURIER (FT-IR)
A espectroscopia é uma técnica baseada na energia absorvida após interação
da radiação com a matéria. Tal interação produz movimento dos átomos que
constituem as moléculas, resultando em rotações e vibrações moleculares.
Basicamente, as vibrações moleculares podem ser classificadas em dois tipos:
vibrações de deformação axial e de deformação angular. As deformações axiais, ou
estiramento, são oscilações radiais das distâncias entre os núcleos enquanto as
deformações angulares envolvem mudanças dos ângulos entre as ligações ou,
como no modo de deformação assimétrica fora do plano, alterações do ângulo entre
o plano que contém as ligações e um plano de referência. Como as ligações
químicas das substâncias possuem frequências de vibração especificas, é possível
identificar uma substancia por tal técnica (SILVERSTEIN; WEBSTER; KIEMLE,
2006).
Assim, essa análise foi realizada com o objetivo de avaliar possíveis
modificações químicas após a recristalização. A molécula A foi caracterizada na
forma original e recristalizada. Todas as amostras foram compactadas com Brometo
de Potássio (KBr), formando pastilhas (99 mg KBr:1 mg molécula). Em seguida,
submeteram-se as pastilhas à análise em um espectrofotômetro da Shimadzu
(modelo IRprestige21) na região de 4000-400 cm-1, 20 varreduras, com resolução de
2 cm-1 na temperatura ambiente.
ANÁLISE TÉRMICA
A análises térmica compreende um grupo de técnicas na qual uma
propriedade física de uma substancia e, ou seus produtos de reação é medida,
enquanto a amostra é submetida a uma programação de temperatura. Dentre as
38
técnicas, encontram-se: a termogravimetria (TG) e a análise térmica diferencial
(DTA). A TG é uma análise que mede a massa de uma amostra em função da
temperatura (IONASHIRO; GIOLITO, 1980). A DTA é uma técnica na qual o
equipamento registra a diferença de temperatura entre uma amostra e uma
substância referência em função da temperatura. Através do DTA é possível
detectar processos que envolvem eventos exotérmicos e endotérmicos (MARTÍNEZ
et al., 2013).
A caracterização por TG e DTA foi realizada para identificação de
propriedades térmicas da molécula A original, como sua temperatura de fusão. As
medidas foram efetuadas em uma termobalança DTG-60 da Shimadzu, em cadinho
de alumínio, massas em torno de 10 mg, na faixa de temperatura de 25°C a 600°C
com a razão de aquecimento de 10°C.min-1 sob atmosfera dinâmica de nitrogênio.
MICROSCOPIA ELETRÔNICA DE VARREDURA
A microscopia eletrônica de varredura (MEV) é uma técnica que possibilita
visualizar a superfície de pequenas partículas através de um aumento de 30000
vezes. O equipamento emite um feixe de elétrons sob a superfície da partícula a ser
analisada. Esses elétrons são retroespalhados ou a superfície do material emite
outros
elétrons.
O
equipamento
analisa
esses
elétrons
secundários
ou
retroespalhados e forma uma imagem, em tons de cinza, da superfície do material
(DEDAVID; GOMES; MACHADO, 2007; DUARTE; JUCHEM; PULZ, 2003).
A morfologia da molécula A na forma original e recristalizada foi analisada por
MEV. As imagens foram obtidas no Laboratório Institucional de Microscopia
Eletrônica de Varredura do Museu Paraense Emílio Goeldi, utilizando-se um
microscópio eletrônico LEO modelo 1450VP. As amostras foram fixadas em fita
dupla face de carbono contidas em um suporte de alumínio de 12 mm de diâmetro.
Em seguida, a fim de se tornarem condutivas, foram metalizadas com Au por 2':30'',
o que deposita sobre a amostra uma película com espessura média de 15 nm. As
imagens foram geradas por detecção de elétrons secundários, utilizando-se
aceleração de voltagem entre 12,5 e 15 kV e distâncias de trabalho variáveis entre
10 e 15 mm.
39
DIFRAÇÃO DE RAIOS-X
A difração de raios-X permite a determinação da estrutura cristalina do sólido.
O sólido é analisado por um feixe de raios-X. A interação dos raios incidentes com a
amostra produz uma interferência construtiva (difração de raios-X). Os raios
difratados são detectados e tratados. A conversão dos picos permite a obtenção de
um espectro de difração (ALBERS; MELCHIADES; MACHADO, 2002). Essa análise
foi realizada para detecção de possíveis mudanças de estado físico e fases
cristalinas após a cristalização. A caracterização da molécula na forma original foi
realizada por Azevedo (2014a). A caracterização das amostras da molécula na
forma recristalizada foi realizada utilizando o difratômetro X PERT-PRO (modelo
PW3050 da PANanalytical), locado no Laboratório de Caracterização Mineral
(Instituto de Geociências–UFPA), (λ = 1,5406 Å) equipado com anodo de Cu, tempo
de contagem 20s, 2θ (5° -75°), corrente de 30 mA e tensão de 40 kV.
4.2
Estudo da solubilidade
O estudo de solubilidade de uma nova molécula é investigado para se
determinar a quantidade da substância que irá se dissolver em dado solvente,
tornando-se como limite o equilíbrio da saturação. Para quantificação real do analito
em estudo foi necessário desenvolver e validar um método analítico por
cromatografia líquida de alta eficiência. Assim, a etapa de validação foi realizada
neste trabalho para garantir a segurança e confiabilidade dos resultados
VALIDAÇÃO DE UM MÉTODO DE QUANTIFICAÇÃO DA MOLÉCULA A EM
SOLUÇÃO POR CROMATOGRAFIA LIQUIDA DE ALTA EFICIÊNCIA (CLAE)
A cromatografia é um método de separação, onde os componentes da
mistura migram de acordo com a interação com a fase estacionária e a fase móvel.
A CLAE se diferência das outras técnicas cromatográficas por utilizar um líquido
40
como fase móvel e por usar uma bomba de alta pressão para eluir a fase móvel pela
fase estacionária. A Figura 6 esquematiza o funcionamento de um cromatógrafo.
Esta técnica é bastante utilizada para separação, identificação e quantificação de
substâncias (DEGANI; CASS; VIEIRA, 2011).
Figura 6 - Esquema básico do funcionamento de um equipamento de CLAE: a reservatório de fase móvel; b - bomba de alta pressão; c - válvula de injeção da
amostra; d - fase estacionária (coluna); e - detector; f – registrador
Para realização das análises o sistema cromatográfico utilizado foi composto
por um cromatógrafo liquido de alta eficiência Varian®, acoplado com uma bomba
isocrática prostar 300, injetor manual reodyne com um detector de canal duplo de
ultravioleta-visível prostar 220 e um sistema de software Star Varian 6.0. As
condições cromatográficas utilizadas foram: coluna Xterra RP18 5 µm (4,6x150mm);
fase móvel composta de uma mistura de acetonitrila:água (1:1); fluxo de 1 mL/min e
volume de injeção de 10µL; comprimento de onda de detecção de 318nm e o tempo
de retenção de 8 minutos.
41
Determinação dos parâmetros de validação
a) Linearidade
É a capacidade da metodologia que mostra o quanto os resultados da técnica
são diretamente proporcionais a concentração do analito em uma determinada faixa
de concentração. A ANVISA (2003) recomenda o uso de pelo menos 5
concentrações diferentes e é considerado linear a metodologia que apresentar, no
mínimo, o coeficiente de correção (r) igual a 0,99.
A linearidade foi avaliada a partir de uma solução estoque feita de 10 mg da
molécula A solubilizada com acetonitrila em um balão de 100 mL, formando uma
solução de concentração igual a 100 µg/mL, chamada de solução mãe (SM). A partir
dessa solução foi preparada as soluções padrões como mostra a Tabela 5 para a
construção de uma curva de calibração.
Tabela 5 - Preparo das soluções para a curva de calibração
Concentração
Volume da
Volume do diluente
Volume
(µg/mL)
SM (mL)
(mL)
final (mL)
1
0,25
0,025
9,975
10
2
0,5
0,050
9,95
10
3
1
0,020
1,98
2
4
2
0,040
1,96
2
5
4
0,080
1,92
2
6
8
0,160
1,84
2
Ponto
Cada ponto foi injetado no sistema cromatográfico e os resultados obtidos
foram plotados em um gráfico da concentração da solução em função da área do
pico. As curvas foram efetuadas em triplicata (n=3), onde obtém-se a curva média e
o seu coeficiente de correlação foi calculado.
42
b) Seletividade
Seletividade é a capacidade da metodologia de medir com exatidão o analito
mesmo na presença de outras substâncias como impurezas, produtos da
degradação e componentes da matriz da amostra em análise (ANVISA, 2003).
Para a dissolução os únicos interferentes na análise foram o glicofurol e o
lauril sulfato de sódio (LSS). Assim a seletividade do método foi avaliada mediante
as leituras
de
uma
solução
de
glicofurol
0,1% diluído na fase
móvel
(acetonitrila/água 1:1) e uma solução de lauril sulfato de sódio 0,5% diluído em água
destilada. Cada solução foi analisada por CLAE e seus cromatogramas foram
comparados com o da solução padrão de 2 µg/mL de molécula A.
c) Precisão
A precisão é a capacidade dos métodos terem resultados próximos em uma
série de medidas de uma amostragem múltipla da mesma amostra. Entre os tipos de
precisão tem-se: a precisão intermediária que é a concordâncias dos resultados no
mesmo laboratório, mas em dias diferente e analistas diferentes; e a repetibilidade
que é a concordância dos resultados no mesmo dia, pelo menos analista. A precisão
pode ser expressa pelo coeficiente de variação (CV%) e seu valor não pode ser
superior a 5% (ANVISA, 2003).
Para avaliar a precisão foi realizado a repetibilidade e a precisão
intermediária. A repetibilidade foi analisada a partir de seis determinações de igual
concentração teórica de 2 µg/mL.
Na precisão intermediária avaliou-se as seguintes concentrações: 0,5 µg/mL,
2 µg/mL e 8 µg/mL. Cada ponto foi analisado em triplicata, por 2 analistas diferentes
e em 2 dias diferentes. Os resultados do parâmetro de precisão foram expressos
através do coeficiente de variação (Equação 11).
𝐶𝑉 (%) =
𝐷𝑒𝑠𝑣𝑖𝑜 𝑝𝑎𝑑𝑟ã𝑜
𝑥100
𝐶𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎çã𝑜 𝑚é𝑑𝑖𝑎 𝑑𝑒𝑡𝑒𝑟𝑚𝑖𝑛𝑎𝑑𝑎
(11)
43
d) Exatidão
A exatidão é a capacidade do método ter seus resultados próximos ao valor
real. Para a análise da exatidão é necessário realizar no mínimo 9 determinações: 3
de uma concentração baixa; 3 de uma média e 3 de uma alta (ANVISA, 2003).
Foram analisadas 3 concentrações: 0,5 µg/mL, 2 µg/mL e 8 µg/mL. De cada
ponto foi realizada uma triplicata e calculada a média desses valores. Com a média
foi possível calcular a recuperação (R) através da Equação 12. Segundo a ANVISA
(2003) esse valor não pode ser menor que 95%.
𝑅(%) =
𝐶𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎çã𝑜 𝑚é𝑑𝑖𝑎 𝑒𝑥𝑝𝑒𝑟𝑖𝑚𝑒𝑛𝑡𝑎𝑙
𝑥 100
𝐶𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎çã𝑜 𝑡𝑒ó𝑟𝑖𝑐𝑎
(12)
e) Limite de detecção e de quantificação
O limite de detecção é o menor valor do analito que pode ser detectado pela
metodologia e o limite de quantificação é o menor valor do analito que pode ser
quantificado com exatidão e precisão (ANVISA, 2003). Os limites de detecção (LD) e
de quantificação (LQ) foram calculados através das Equações 13 e 14
respectivamente.
𝐿𝐷 =
𝐿𝑄 =
𝐷𝑃𝑎 𝑥 3
𝐼𝐶
𝐷𝑃𝑎 𝑥 10
𝐼𝐶
(13)
(14)
Onde, DPa é o desvio padrão do intercepto do eixo y em 3 curvas de
calibração e IC é a inclinação da curva de calibração.
44
SOLUBILIDADE APARENTE DA MOLÉCULA A EM GLICOFUROL E EM
MISTURAS DE GLICOFUROL/ÁGUA
Em um Becker de 25 mL foi adicionado 10 mL de glicofurol e em outros 3
béckeres foram preparadas 3 misturas de glicofurol/água em diferentes proporções
como mostra a Tabela 6. Em seguida, sob agitação magnética, foram adicionadas
amostras de massas conhecidas de molécula A nas soluções. Foram realizadas as
adições até que a solução estivesse viscosa o suficiente que não permitisse a
adição de mais sólido.
Tabela 6 - Preparação das soluções em diferentes proporções de glicofurol/água
Volume de glicofurol
Volume de água
Volume final
Glicofurol/água (1:2)
5 mL
10 mL
15 mL
Glicofurol/água (1:1)
7,5 mL
7,5 mL
15 mL
Glicofurol/água (2:1)
10 mL
5 mL
15 mL
Ao término da adição de sólido nas soluções, os quatro béckeres foram
colocados em banho-maria a 25°C por 72 horas. Cada dia os béckeres foram
retirados do banho e submetidos a agitação magnética. Este procedimento foi
realizado três vezes a cada dia. Posteriormente as misturas foram centrifugadas
sendo o sobrenadante coletado para a quantificação em CLAE da molécula A que
ficou solúvel. A parte solida (não solúvel) do ensaio foi colocado em uma placa de
petri e posto em estufa a 120°C, após 24 horas o material foi levado ao dessecador
para esfriar até temperatura ambiente e ser pesado em balança analítica, voltando
para a estufa novamente. O procedimento foi repetido até a amostra atingir peso
constante. Através da determinação do peso final, tem-se a massa de molécula A
não solubilizada e pela subtração do total que foi utilizado, calcula-se o quanto de
molécula A foi solubilizada, o que corresponde à sua solubilidade aparente.
45
Esta análise não mostra um valor exato da solubilidade da molécula A, mas
ela possibilita o cálculo do fator de diluição para outras análises como a
cromatografia líquida de alta eficiência também usada nesse trabalho.
SOLUBILIDADE “REAL” DA MOLÉCULA A EM GLICOFUROL E EM
MISTURAS DE GLICOFUROL/ÁGUA
Após a validação da metodologia de quantificação por CLAE foi possível
quantificar a massa real de molécula A solubilizada em glicofurol e nas misturas de
glicofurol/água.
4.3
Preparação de cristais através da preciptação por anti-solvente
ENSAIO PRELIMINAR DE CRISTALIZAÇÃO
Em um tubo cônico contendo 0,5 mL de água ultra pura foram gotejados em
0,5 mL de solução saturada de molécula A com o auxílio de uma pipeta de pasteur.
A cada duas gotas, a solução foi agitada em um agitador tipo vortex. A mistura ficou
em repouso por 24 horas para a formação dos cristais. Os cristais foram coletados
com auxilio de uma pipeta de pasteur e analisados em um microscópio óptico
comum, nas objetivas de 4x, 10x e 40x.
PREPARAÇÃO DOS CRISTAIS
Depois do teste preliminar de recristalização, novos testes foram realizados
utilizando-se diferentes quantidades de anti-solvente (água) em relação à
quantidade de solução de molécula A em glicofurol, como mostra a Tabela 7.
46
Tabela 7 - Proporção de solução saturada e água para a recristalização.
Proporção
Volume de Solução
Volume
Volume final
Saturada (mL)
de água
Solução/água (1:2)
5
10
15
Solução/água (1:1)
7,5
7,5
15
Solução/água (2:1)
10
5
15
Para cada uma das preparações procedeu-se da seguinte forma: Um becker
com água permanecia em um agitador magnético com velocidade de 300 rpm; Com
o auxílio de uma pipeta de pasteur gotejava-se lentamente a solução de molécula A.
Em seguida, a mistura era filtrada e lavada com água para a retirada do excesso de
solvente. A parte sólida era separada e colocada para secar na estufa a 120°C.
DETERMINAÇÃO DA MELHOR PROPORÇÃO ÁGUA/GLICOFUROL PARA
A RECRISTALIZAÇÃO DA MOLÉCULA A
Com base na solubilidade da molécula A em glicofurol e em misturas de
glicofurol e água pode-se calcular o rendimento em produto (massa de cristais
formados). Para calcular o rendimento da produção de cristais aplicou-se a Equação
15.
Rdt 1
Srx(1 Rx)
Sgly
(15)
Onde,
Rdt é o rendimento,
Srx é a solubilidade (g/mL) da molécula A na mistura de glicofurol/água em
análise,
Sgly é a solubilidade de molécula A (g/mL) em glicofurol,
Rx é a razão da quantidade de água por glicofurol na mistura em análise.
47
4.4
Caracterização do produto recristalizado
Os cristais obtidos produto recristalizado foi analisado por FT-IR, DRX e MEV,
em comparação à molécula original.
PERFIL DE DISSOLUÇÃO
Os perfis de dissolução da molécula original e dos produtos recristalizados
foram determinados utilizando-se o dissolutor SOTAX® AT 7smart Manual.
Dois meios de dissolução foram testados :
- Água destilada com pH=7 ;
- Solução aquosa contendo LSS 0,5%, pH=7,4. Quando a solubilidade do
fármaco
em água é baixa recomenda-se utilizar o LSS como meio
alternativo (FDA, 1997).
Foram utilizadas 3 cubas para cada meio. Em cada uma delas, colocou-se 14
mg da amostra em teste para um volume de meio de 900 mL. As demais condições
mantidas : rotação das pás de 50 rpm; temperatura de 37°C; tempos de coleta da
amostra de 1, 5, 10, 20, 30 e 60 minutos. A quantificação foi realizada por CLAE.
Os dados foram tratados no software sigmaplot procurando o melhor modelo
cinético para representar o perfil de dissolução. Os modelos testados foram: Primeira
ordem ; Higuchi ; Hixon e Crowell ; Peppas ; e Baker e Londsdale.
Os perfis de dissolução da molécula A e de seu recristalizado foram
comparados através do calculo da eficácia de dissolução, do fator de similaridade e
do fator de diferença.
48
5
5.1
RESULTADOS E DISCUSSÃO
Caracterização da molécula A
ESPECTROMETRIA DE INFRAVERMELHO COM TRANSFORMADAS DE
FOURIER (FT-IR)
A Figura 7 mostra o espectro FT-IR da molécula A. Observa-se um pico em
3178 cm-1 atribuido a associação de amida com amina. Em 3019 cm -1 tem-se um
pico característico de deformação axial de C-H aromático. Em 1654 cm-1 há um pico
forte que corresponde a carbonila de amida. Na região de 1133 cm-1 evidencia-se
uma deformação axial assimétrica de C-O-C. Na faixa de 1250-1020 cm-1 existem
picos caracteristicos da absorção de grupos tiocarbonila (C=S). Uma banda larga na
faixa de 800-666 cm-1 (693 cm-1) é causada pela deformação angular simétrica fora
do plano do grupo N-H (SILVERSTEIN; WEBSTER; KIEMLE, 2006).
Figura 7 - Espectro de infravermelho da molécula A
49
Os grupos funcionais que foram evidenciados pela avaliação da FT-IR
corroboram com a estrutura do cardiotônico em estudo como mostra a Figura 8. A
molécula A de acordo com a nomenclatura IUPAC corresponde à N-[(E)-thiophen-2ylmethylidene]-1,3-benzodioxole-5-carbohydrazide.
Figura 8 - Forma estrutural da molécula A.
ANALISE TÉRMICA
A análise térmica da molécula A mostra um pico endotérmico à temperatura
onset de 205,4°C (Figura 9). Picos endotérmicos são causados por fusão,
vaporização, sublimação, dessorção, absorção, transição de cristal líquido, entre
outros (BERNAL et al., 2002). Como este foi o único evento térmico observado,
supõe-se que seja o ponto de fusão da molécula A.
Figura 9 - TG e DTA da molécula A
50
Azevedo (2014b), em seu estudo, utilizou 2 mg de molécula a e encontrou o
valor de 205°C como ponto de fusão. Apesar de se ter usado 10 mg o ponto de
fusão não apresentou variação, mostrando que o calor se distribuiu uniformemente
na amostra (BERNAL et al., 2002).
MICROSCOPIA ELETRÔNICA DE VARREDURA
A Figura 10 apresenta uma fotomicrografias obtida por MEV da molécula A.
Observa-se que a molécula A na sua forma original apresenta-se em forma de
cristais de tamanhos variados. Esta micrografia será utilizada para estudo
comparativo da molécula A com seus recristalizados.
Figura 10 - Microscopia eletrônica de varredura da molécula A em diferentes
magnitudes A (121x), B(316x), C(450x), D(1270x).
51
5.2
Estudo da solubilidade
VALIDAÇÃO DO MÉTODO DE QUANTIFICAÇÃO DA MOLÉCULA A POR
CLAE
a) Linearidade
A linearidade do método analítico foi analisada através da média das
curvas analíticas (Tabela 8). O gráfico plotado concentração versus área obteve
um coeficiente de correlação linear de 0,9996, significando 99,96% da variação
total em torno da média (Figura 11). O critério mínimo aceitável para o coeficiente
de correlação deve ser igual a 0,99 (ANVISA, 2003).
Tabela 8 - Curvas de calibração preparadas em 3 dias diferentes.
Concentração
Curva 1
Curva 2
Curva 3
(µg/mL)
(mV/s2)
(mV/s2)
(mV/s2)
0,25
5,52
6,80
0,50
16,40
1
Média
Desvio Padrão
9,55
7,29
2,06
15,70
18,10
16,73
1,23
46,90
37,80
44,80
43,17
4,76
2
81,00
86,30
84,20
83,83
2,67
4
199,00
169,00
177,00
181,67
15,53
8
338,00
364,00
367,00
356,33
15,95
r²
0,9902
0,9991
0,9995
0,9996
Área (mV/s2)
52
400
350
300
250
200
150
100
50
0
0,00
y = 45,252x - 3,949
R² = 0,9996
2,00
4,00
6,00
8,00
Concentração (µg/mL)
10,00
Figura 11 - Curva de calibração correlacionado a Área dos picos com a concentração
b) Seletividade
A seletividade foi avaliada para observar o grau de interferência das soluções
na determinação da molécula A. A Figura 12 apresenta o cromatograma do LSS
(0,5%) e do glicofurol (1%), comparados com a molécula A (2 µg/mL), a qual ilustra
que não houve interferência das soluções de LSS e glicofurol, o que garante o pico
de resposta exclusivo da molécula A no tempo de retenção próximo a 4,7 minutos.
Figura 12 - Cromatogramas do Lauril sulfato de sódio 0,5% (verde), glicofurol a 0,1%
(vermelho) e molécula A a 2 µg/mL (azul)
53
c) Precisão
Repetibilidade
A repetibilidade foi determinada através de seis análises da amostra de
concentração 2 µg/mL (Tabela 9). O Coeficiente de variação foi de 2,59% mostrando
que o método é preciso e está dentro da faixa que é preconizado pela ANVISA
(2003), que determina o coeficiente de variação menor que 5%.
Tabela 9 - Repetibilidade do padrão de concetração de 2 µg/mL
Área
Concentração
(mV/s2)
(µg/mL)
93,5
88,2
2,07
1,95
88,0
1,95
90,6
2,01
93,2
2,06
91,7
2,03
Média
2,01
Desvio padrão
0,05
Coeficiente de variação (%)
2,59
Precisão intermediária
Os efeitos das variações do método dentro do mesmo laboratório foram
observados através da precisão intermediária. A Tabela 10 apresenta os valores da
precisão intermediária e seu coeficiente de variação. Todos os coeficientes estão
abaixo dos 5%, valor aceito pela ANVISA (2003), mostrando que o método é
preciso.
54
Tabela 10 - Resultado da precisão intermediaria
dia1
0,5
2
8
0,55
2,18
8,67
0,54
2,06
7,88
0,52
2,11
8,31
0,54
2,12
8,29
0,02
0,06
0,39
Coeficiente
de
variação
2,93
2,82
4,75
dia 2
0,5
2
8
0,55
2,30
8,89
0,54
2,31
8,26
0,59
2,38
8,40
0,56
2,33
8,52
0,03
0,04
0,33
4,38
1,82
3,89
dia 1
0,5
2
8
0,55
2,42
8,62
0,58
2,38
8,94
0,58
2,28
8,53
0,57
2,36
8,70
0,02
0,07
0,21
2,87
2,96
2,44
dia 2
0,5
2
8
0,53
2,24
8,02
0,52
2,11
8,20
0,55
2,15
7,99
0,53
2,17
8,07
0,01
0,07
0,11
2,54
3,27
1,37
Concentração
(µg/mL)
Média
Desvio
padrão
Analista 1
Analista 2
d) Exatidão
Através da exatidão calculou-se o grau de concordância entre os resultados
experimentais e o valor teórico aceito como referência. A Tabela 11 mostra os
valores encontrados das análises em CLAE para as concentrações baixa, média e
alta, junto com o resultado das recuperações. Os valores estão dentro do
determinado pela ANVISA (2003) mostrando que o método é exato.
Tabela 11 - Dados da recuperação do método
0,5 µg/mL
2 µg/mL
8 µg/mL
0,51
2,12
6,51
0,55
2,06
8,82
0,51
2,02
7,79
Média
0,52
2,07
7,71
Recuperação (%)
103,68
103,43
96,35
55
e) Limite de detecção e Limite de quantificação
Os interceptos do eixo Y das 3 curvas foram: 0,1072; -7,5692; e -2,6139. O
desvio padrão (DPa) é 3,892. A inclinação da curva (IC), retirada da equação da
curva de calibração usada na linearidade, é de 45,252. Usando as formulas
apresentadas na metodologia o limite de detecção é de 0,258 µg/mL e o limite de
quantificação é de 0,86 µg/mL.
SOLUBILIDADE APARENTE
A solubilidade aparente da molécula A em glicofurol foi realizada através de 4
adições do sólido em 10 mL (10625,3 mg) do solvente, sob agitação. A Tabela 12
apresenta os valores adicionados.
Tabela 12 - Adição de molécula A em glicofurol.
Número de adições
1
2
3
4
Total
Molécula A (mg)
100,1
105,3
97,0
128,5
430,9
Depois da 4° adição, formou-se um corpo de fundo e esse foi separado por
centrifugação como proposto pela metodologia. A parte não solubilizada teve uma
massa de 135,2 mg. Dessa forma, a massa de molécula A presente em 10625,3 mg
glicofurol foi de 295,3 mg e a solubilidade aparente da molécula A em glicofurol é de
27,83 mg de molécula A a cada 1000 mg de glicofurol.
A solubilidade da molécula A em misturas de glicofurol e água foi determinada
de forma similar à calculada para a solubilidade em glicofurol. Os dados da
solubilidade aparente da molécula A em diferentes misturas de glicofurol e água
estão apresentados na Tabela 13.
56
Tabela 13 - Solubilidade da molécula A em misturas de glicofurol e água.
Massa de molécula A (mg)
Proporção
Concentração (mg/mL)
solubilizada em 15 mL
Glicofurol/água (1:2)
105
7
Glicofurol/água (1:1)
99,5
6,6
Glicofurol/água (2:1)
198,7
13,2
SOLUBILIDADE
REAL
DA
MOLÉCULA
A
EM
MISTURAS
DE
GLICOFUROL/ÁGUA
Uma vez validada a metodologia de quantificação por CLAE, foi possível
determinar a solubilidade "real" da molécula A em glicofurol e nas misturas de
glicofurol/água. Os resultados são apresentados na tabela abaixo:
Tabela 14 - Quantificação da molécula A em diferentes meios saturados
Proporção solvente
Concentração(mg/mL)
Glicofurol
32,31
Glicofurol/água (1:2)
0,45
Glicofurol/água (1:1)
1,43
Glicofurol/água (2:1)
8,60
De acordo com resultados apresentados na Tabela 14, a solubilidade da
molécula A em glicofurol foi de cerca de 32 mg/mL. Essa solubilidade é da mesma
ordem de grandeza da solubilidade de outras moléculas farmacêuticas em glicofurol,
como a carbamazepina (100 mg/mL) (Tauboll et al, 1990). O uso do glicofurol neste
estudo de recristalização da molécula A justifica-se por causa da sua baixa
toxicidade (ALLHENN; LAMPRECHT, 2011).
57
5.3
Preparação de cristais através da cristalização da molécula A em mistura
glicofurol/água
ENSAIO DE CRISTALIZAÇÃO
A microscopia óptica (Figura 13) mostra que ocorreu a formação de cristais
quando a solução foi adicionada na água, o que justifica que ela pode ser utilizada
para a preparação de cristais através da técnica de recristalização por anti-solvente.
.
Figura 13 - Microscopia optica do recristalizado de molécula com água utilizando
glicofurol como solvente
58
DETERMINAÇÃO DA MELHOR PROPORÇÃO GLICOFUROL/ÁGUA PARA
A RECRISTALIZAÇÃO DA MOLÉCULA A
A Tabela 15 mostra os valores de solubilidade da molécula A em diferentes
misturas de glicofurol e água (Srx), a razão da quantidade de água por glicofurol
(Rx) e o rendimento da produção de cristais (Rdt) calculada, sabendo que a
solubilidade de molécula A me glicofurol (Rgly) é 32,31 mg/mL.
Solubilidade (mg/mL)
Tabela 15 - Cálculo do rendimento da produção de cristais
Proporção
Srx (g/mL)
Rx
Rdt (%)
Glicofurol/água (1:2)
0,45
2
95,82
Glicofurol/água (1:1)
1,43
1
91,16
Glicofurol/água (2:1)
8,60
0,5
60,43
10
9
8
7
6
5
4
3
2
1
0
0,5
1
Razão água/glycofurol
2
Figura 14 - Gráfico da solubilidade de molécula A em diferentes misturas de
glicofurol/água.
59
Através da Figura 14 observa-se que quanto maior a quantidade de água
(anti-solvente) menor é a solubilidade da molécula A e isso é comprovado no cálculo
do rendimento onde a proporção de quantidade de água (proporção 1-2) é a de
maior rendimento. Assim, essa é a melhor proporção utilizada para a produção de
cristais.
5.4
Caracterização da molécula A e de seus recristalizados
INFRAVERMELHO COM TRANFORMADA DE FOURIER
A molécula A e seus recristalizados apresentam o mesmo espectro de
infravemelho como mostra a Figura 15. Este resultado evidencia que o processo de
recristalização estudado não alterou a estrutura química da molécula A.
Figura 15 - Espectros de infravermelho da molécula A (preto) e de seus recristalizados
na proporção de 2-1 (vermelho), 1-1 (azul) e 1-2 (rosa)
60
DIFRAÇÃO DE RAIO-X
Através da difração de raio-x (Figura 16) observa-se que tanto a molécula A
como seus recristalizados apresentam picos ao longo de todo o difratograma
confirmando suas estruturas cristalinas. Observa-se também não houve a formação
de novas formas cristalinas (não aparecimento de novos picos).
Figura 16 - Difração de raios-x da molécula A (preto) e de seus recristalizado na
proporção de 2-1 (vermelho), 1-1 (azul) e 1-2 (rosa)
MICROSCOPIA ELETRÔNICA DE VARREDURA
As imagens obtidas da molécula A na forma recristalizada, na proporção de 21, 1-1 e 1-2 são mostradas respectivamente nas Figura 17, Figura 18 e Figura 19.
61
Figura 17 - Microscopia eletrônica de varredura do recristalizado na proporção 2-1 nas
seguintes magnitudes: A(100x), B(2080x), C (12450x), D (13960x)
.
Figura 18 - Microscopia eletrônica de varredura do recristalizado na proporção 1-1 nas
seguintes magnitudes: A(80x), B(3360x), C (1300x), D (10170x)
62
Figura 19 - Microscopia eletrônica de varredura do recristalizado na proporção 1:2 nas
seguintes magnitudes: A(196x), B(621x), C (3870x), D (10710x)
Através das imagens, nota-se que os recristalizados encontram-se bastante
agregados e apresentam pequenos cristais em torno de 3 µm. Essa redução de
tamanho e modificação de morfologia (cristais mais finos e alongados) pode ser
comprovada na Figura 20 que compara os cristais da molécula A forma original e
recristalizada na magnitude de 1270x.
Figura 20 - Microscopia eletrônica de varredura da molécula A original (A) e
recristalizada na proporção de 1-1 (B) na magnitude de 1270x.
63
DISSOLUÇÃO
Realizou-se a dissolução in vitro da molécula A na forma original e
recristalizada na proporção 1-2 (ensaio que apresentou melhor rendimento). Os
dados obtidos são apresentados na Tabela 16, para os dois diferentes meios de
dissolução utilizados.
Através dos perfis de dissolução, testou-se 5 modelos cinéticos. De acordo
com as equações matemáticas, o modelo que melhor explica o perfil de dissolução é
aquele onde o coeficiente de correlação de Pearson (r2) mais se aproxima de 1.
O modelo cinético que melhor explicaria o perfil de dissolução da molécula A
em água é o modelo de Hixon e Crowell (dissolução de cristais com variação de
área superficial), porém seu r² foi muito baixo não sendo o suficiente para explicar o
perfil cinético do fármaco. Isso ocorreu pelo fato de apenas ter sido possível
quantificar por CLAE a concentração do fármaco dissolvido após 60 minutos de
ensaio (último ponto de amostragem, Tabela 16).
Tabela 16 - Perfil de dissolução da molécula A em água e em LSS 0,5%
Quantidade dissolvida da Molécula
Quantidade dissolvida da Molécula
Tempo
A na forma original
A na forma recristalizada
(min)
(% ± DP)
(% ± DP)
Agua
LSS
Agua
LSS
1
0,00
6,63 ± 0,12
0,00
4,22 ± 0,26
5
0,00
17,30 ± 0,24
0,00
7,40 ± 0,16
10
0,00
27,74 ± 0,63
0,00
8,12 ± 0,4
20
0,00
44,56 ± 1,77
0,00
13,46 ± 0,03
30
0,00
58,79 ± 2,02
0,18 ± 0
14,18 ± 0,3
60
1,63±0,03
68,43 ± 2,27
2,00 ± 0,02
19,77 ± 0,38
64
Utilizando os valores da Tabela 16 pode-se comparar a performance em
dissolução da molécula A na forma original e recristalizada. Como mostra a Figura
21, os valores de dissolução em água foram muito baixos.
Concentação (%)
2,5
2,0
1,5
1,0
0,5
0,0
0
10
20
30
40
Tempo (minutos)
Molecula A em agua
50
60
70
Recristalizado em água
Figura 21 - Perfil de dissolução da molécula A (vermelho) e de seu recristalizado (azul)
usando como meio água
A Figura 22 apresenta o perfil de dissolução da molécula A e de seu
recristalizado utilizando LSS como meio de dissolução. Pode-se observar que a
molécula A possui uma melhor performance do que seu recristalizado. Para
comprovar isso foram realizados os cálculos de fator de semelhança e de
similaridade, para saber se os perfis são diferentes, e eficácia de dissolução, para
saber qual possui a melhor performance.
65
Concentração (%)
80
70
60
50
40
30
20
10
0
0
10
20
30
40
Tempo minutos
Molecula A em LSS
50
60
70
Recristalizado em LSS
Figura 22 - Perfil de dissolução da Molécula A (azul) e de seu recristalizado (vermelho)
usando como meio LSS 0,5%.
O resultado encontrado para F1 foi de 69,95 e o F2 foi de 25,33. Para se ter
semelhança, o F1 tem que ser próximo a 1 sendo aceitável valores menores que 15
e o F2 tem que ser próximo a 100 sendo aceitável valores acima de 50. Nesse caso
Como o F1 foi mais de 15 e o F2 menor que 50, as curvas são diferentes.
Através da eficácia de dissolução (Tabela 17) pode-se observar que a
molécula A apresenta uma melhor performance para a dissolução que o seu
recristalizado. O recristalizado, por possuir partículas menores deveria ter uma
melhor eficácia de dissolução, porém a existência de aglomerados parece ter
dificultado a dissolução do recristalizado (YOUN et al., 2011). Para contornar esse
problema alguns autores utilizam sufactantes como a lactose (ALLAHHAM;
STEWART; DAS, 2013; STEWART; ZHAO, 2005) ou então, para diminuir a
agregação das partículas, seca-se os cristais por “spray drying” ao invés de uma
secagem em estufa que favorece a aglomeração (POUTON, 2006).
66
Tabela 17 - Cálculo da eficácia de dissolução
Área total
Área da curva
ED (%)
Molécula A em LSS
6000
2,946.89
49.11
Recristalizado em LSS
6000
817.35
13.62
Os cálculos dos r2 dos modelos matemáticos das dissoluções são
apresentados na Tabela 18. Os modelos que melhor explicaram o perfil cinético do
Molécula A em LSS 0,5% foram o modelo de Higuchi, modelo de baker e lonsdale e
o modelo de Peppas. Para a molécula na forma recristalizada, o modelo cinético que
melhor se aplicou foi o de Peppas (Tabela 18). No primeiro caso a dissolução ocorre
apenas pela difusão por isso os 3 modelos justificam a curva, porém no segundo
caso além da difusão ocorre também a desagregação das partículas por isso o
modelo de Peppas é o que mais justifica a curva pois é um modelo aplicado quando
a dissolução ocorre por mais de um tipo de liberação (COSTA, 2002). Nas Figuras
23, 24 e 25 podendo ser observados o perfil de dissolução da molécula A em LSS
0,5%, do recristalizado em água e do recristalizado em LSS 0,5% com a linha de
tendência do modelo matemático de Peppas, respectivamente.
Tabela 18 - Cálculo dos r² de alguns modelos matemáticos
Hixon e
Higuchi
Molécula A em água
0,66
0,37
0,66
--
0,37
Molécula A em LSS
0,93
0,96
0,86
0,96
0,96
Recristalizado em água
0,72
0,42
0,72
1,00
0,42
Recristalizado em LSS
0,40
0,95
0,37
0,98
0,96
Crowell
Peppas
Baker e
1° ordem
Lonsdale
67
Molecula A em LSS 0,5% no modelo Peppas
F=k*t^n
70
60
Concentração (%)
50
40
30
20
10
0
0
20
40
60
Tempo (min)
Recristalizado
em LSS
no modelo
Peppas
Figura 23 - Molécula
A em LSS
0,5%0,5%
no modelo
Peppas
F=k*t^n
F=k*t^n
2,5
Concentração (%)
2,0
1,5
1,0
0,5
0,0
0
20
40
60
Tempo (min)
Figura 24 - Recristalizado em água no modelo Peppas F=k*t^n
68
Recristalizado em LSS 0,5% no modelo Peppas
F=k*t^n
22
20
18
Concentração (%)
16
14
12
10
8
6
4
2
0
20
40
60
Tempo (min)
Figura 25 - Recristalizado em LSS 0,5% no modelo Peppas F=k*t^n
69
6
CONCLUSÃO
Através de uma
técnica de recristalização por anti-solvente foi possível
preparar cristais da molécula A utilizando-se o glicofurol como solvente e água como
anti-solvente. O método de doseamento por cromatografia líquida de alta eficiência
desenvolvido mostrou-se seletivo, linear, preciso e exato segundo as especificações
da ANVISA (2003) para determinação da concentração da molécula em solução. O
trabalho também necessitou da realização de uma etapa de caracterização físicoquímica da molécula, cujas propriedades ainda são pouco conhecidas e não estão
disponiveis na literatura. Os cristais obtidos da molécula A, através da tecnologia
proposta, apresentaram a mesma estrutura cristalina e foram menores que os
cristais da molécula na forma original. Entretanto, o efeito esperado para otimizar a
cinética de dissolução da molécula não foi verificado, devido a aglomeração dos
cristais obtidos. Embora esses resultados não tenham satisfatórios neste estudo, o
processo de cristalização usando o glicofurol, solvente de baixa toxicidade, mostrouse interessante por manter a forma cristalina do fármaco e, assim, ele pode
continuar sendo uma alternativa para cristalização de outros tipos de fármacos de
baixa solubilidade.
70
7
PERSPECTIVAS E SUGESTÃO DE CONTINUIDADE
Algumas sugestões para continuidade do trabalho:
-
Aprofundar o estudo experimental do processo, introduzindo novas
variáveis de estudo, como por exemplo o modo de mistura da solução com
o anti-solvente, afim de reduzir o problema da aglomeração dos cristais
formados e evidenciar o efeito positivo da redução do tamanho do produto
recristalizado;
-
Introduzir aditivos no anti-solvente (tensoativos ou polímeros hidrofílicos
como HPMC) ou melhorar o método de secagem (secagem por spray
drying);
-
Estudar o efeito dessas novas variáveis na cinética de dissolução;
-
Aprofundar à análise dos modelos cinéticos afim de propor um mecanismo
de dissolução para os cristais formados.
71
8
REFERÊNCIAS
AALTONEN, J. et al. Solid form screening: a review. European journal of
pharmaceutics and biopharmaceutics, v. 71, n. 1, p. 23–37, jan. 2009.
ALBERS, A.; MELCHIADES, F.; MACHADO, R. Um método simples de
caracterização de argilominerais por difração de raios X (A simple method for the
characterization of clay minerals by X-ray diffraction). Cerâmica, v. 48, n. 1, p. 34–
37, 2002.
ALLAHHAM, A.; STEWART, P. J.; DAS, S. C. Improving the de-agglomeration and
dissolution of a poorly water soluble drug by decreasing the agglomerate strength of
the cohesive powder. International journal of pharmaceutics, v. 457, n. 1, p. 101–
9, 30 nov. 2013.
ALLHENN, D.; LAMPRECHT, A. Microsphere preparation using the untoxic solvent
glycofurol. Pharmaceutical research, v. 28, n. 3, p. 563–71, mar. 2011.
ALVES-SILVA, I.; SÁ-BARRETO, L. Preformulation studies of itraconazole
associated with benznidazole and pharmaceutical excipients. Thermochimica Acta,
v. 5, p. 0–4, 2014.
AMIDON, G. L. et al. A theoretical basis for a biopharmaceutic drug classification: the
correlation of in vitro drug product dissolution and in vivo bioavailability.
Pharmaceutical research, v. 12, n. 3, p. 413–20, mar. 1995.
ANVISA. Resolução - RE no 899, de 29 de maio de 2003. Guia para validação de
métodos analíticos e bioanalíticos.
ARAUJO, G.; JR, A. P. Polimorfismo na produção de medicamentos. Revista de
Ciência Farmaceuticas Básicas e Aplicada, v. 33, n. 1, p. 27–36, 2012.
ATTWOOD, D.; FLORENCE, A. T. Princípios físico-químicos em farmácia. [s.l.]
Pharmabooks, 2003.
AUBERT-POUËSSEL, A. et al. Preparation of PLGA microparticles by an emulsionextraction process using glycofurol as polymer solvent. Pharmaceutical research, v.
21, n. 12, p. 2384–91, dez. 2004.
AZEVEDO, J. R. DE. Solubility of a New Cardioactive Prototype Drug in Ionic
Liquids. Journal of Chemical & Engineering Data, v. 59, n. 6, p. 1766–1773,
2014a.
AZEVEDO, J. R. DE. Étude de la cristallisation d’une nouvelle molécule à
efficacité cardiotonique dans un mélange liquide ionique-eau. [s.l.] UNIVERSITÉ
DE TOULOUSE, 2014b.
72
BAGGER, M. A.; NIELSEN, H. W.; BECHGAARD, E. Nasal bioavailability of peptide
T in rabbits: Absorption enhancement by sodium glycocholate and glycofurol.
European Journal of Pharmaceutical Sciences, v. 14, p. 69–74, 2001.
BARAKAT, N. S. Evaluation of glycofurol-based gel as a new vehicle for topical
application of naproxen. AAPS PharmSciTech, v. 11, n. 3, p. 1138–46, set. 2010.
BARREIRO, E. Estratégia de simplificação molecular no planejamento racional de
fármacos: a descoberta de novo agente cardioativo. Química Nova, v. 25, n. 6, p.
1172–1180, 2002.
BERNAL, C. et al. Influência de alguns parâmetros experimentais nos resultados de
análises calorimétricas diferenciais-DSC. Química Nova, v. 25, n. 5, p. 849–855,
2002.
BOONGIRD, A. et al. Biocompatibility study of glycofurol in rat brains. Experimental
biology and medicine, v. 236, n. 1, p. 77–83, jan. 2011.
BRANDÃO, A. L. A. Influência do polimorfismo na farmacotécnica de capsulas
no
setor
magistral.
Disponível
em:
<http://www.intecq.com.br/files/artigos/polimorfismo_e_farmacocinetica.pdf>. Acesso
em: 11 nov. 2014.
BRASIL. Farmacopeia Brasileira. 5° ediçao ed. Brasilia: Anvisa, 2010a. v. 1
BRASIL. Resolução da Diretoria Colegiada - RDC No 31, DE 11 DE AGOSTO DE
2010 da Agência Nacional de Vigilância Sanitária. Dispõe sobre a realização
dos Estudos de Equivalência Farmacêutica e de Perfil de Dissolução
Comparativo. Diário Oficial da União, Brasília: [s.n.].
BROWN, C. K. et al. Acceptable Analytical Practices for Soluble Compounds.
Pharmaceutical Technology, n. December 2004, p. 56–65, 2004.
BURY, R. W. et al. Disposition of intravenous glycofurol: effect of hepatic cirrhosis.
Clinical pharmacology and therapeutics, v. 36, n. 1, p. 82–4, jul. 1984.
CHORILLI, M.; SOUZA, A. Estudo de perfil de dissolução dos medicamentos de
referência, genérico e similar contendo cefalexina na forma farmacêutica cápsula.
Revista de Ciências Farmacêuticas Básica e Aplicada, v. 31, n. 1, p. 69–73,
2010.
COSTA, P. DA. Avaliação in vitro da lioequivalência de formulações farmacêuticas.
Revista Brasileira de Ciências Farmacêuticas, v. 38, 2002.
COSTA, P. An alternative method to the evaluation of similarity factor in dissolution
testing. International journal of pharmaceutics, v. 220, n. 1-2, p. 77–83, 4 jun.
2001.
COSTA, P.; SOUSA LOBO, J. M. Modeling and comparison of dissolution profiles.
European Journal of Pharmaceutical Sciences, v. 13, n. 2, p. 123–33, maio 2001.
73
DEDAVID, B. A.; GOMES, C. I.; MACHADO, G. Microscopia eletrônica de
varredura: aplicações e preparação de amostras: materiais poliméricos,
metálicos e semicondutores. Porto Alegre: EDIPUCRS, 2007.
DEGANI, A. L. G.; CASS, Q. B.; VIEIRA, P. C. Cromatografia um breve ensaio.
Química Nova na Escola, v. 7, 2011.
DHIRENDRA, K. et al. Solid dispersions: a review. Pakistan journal of
Pharmaceutical Sciences, v. 22, n. 2, p. 234–246, 2009.
DONG, Y. et al. Preparation and characterization of spironolactone nanoparticles by
antisolvent precipitation. International journal of pharmaceutics, v. 375, p. 84–88,
2009.
DUARTE, L.; JUCHEM, P.; PULZ, G. Aplicações de microcospia eletrônica de
varredura (MEV) e sistema de energia dispersiva (EDS) no estudo de gemas
exemplos brasileiros. Pesquisas em Geociências, v. 30, n. 2, p. 3–15, 2003.
DURIG, T.; FASSIHI, A. R. Preformulation study of moisture effect on the physical
stability of pyridoxal hydrochloride. International Journal of Pharmaceutics, v. 77,
n. 2-3, p. 315–319, nov. 1991.
ELIAZ, R. E.; SZOKA, F. C. Robust and prolonged gene expression from injectable
polymeric implants. Gene therapy, v. 9, p. 1230–1237, 2002.
FDA. Guidance for Industry Guidance for Industry Dissolution Testing of Immediate
Realise Solid Oral Dosage Forms. n. August, 1997.
GUO, Y.; SHALAEV, E.; SMITH, S. Physical stability of pharmaceutical formulations:
solid-state characterization of amorphous dispersions. Trends in Analytical
Chemistry, v. 49, p. 137–144, set. 2013.
IVATURI, V. D. et al. Bioavailability and tolerability of intranasal diazepam in healthy
adult volunteers. Epilepsy Research, v. 84, p. 120–126, 2009.
JINNO, J. et al. Effect of particle size reduction on dissolution and oral absorption of
a poorly water-soluble drug, cilostazol, in beagle dogs. Journal of controlled
release, v. 111, n. 1-2, p. 56–64, 10 mar. 2006.
KAKRAN, M. et al. Fabrication of quercetin nanoparticles by anti-solvent precipitation
method for enhanced dissolution. Powder Technology, v. 223, p. 59–64, jun. 2012.
KAWABATA, Y. et al. Formulation design for poorly water-soluble drugs based on
biopharmaceutics classification system: basic approaches and practical applications.
International journal of pharmaceutics, v. 420, n. 1, p. 1–10, 25 nov. 2011.
KOVAČIČ, N. N. et al. Influence of the physiological variability of fasted gastric pH
and tablet retention time on the variability of in vitro dissolution and simulated plasma
profiles. International journal of pharmaceutics, v. 473, n. 1-2, p. 552–559, 24 jul.
2014.
74
KUMAR, V.; WANG, L.; RIEBE, M. Formulation and stability of itraconazole and
odanacatib
nanoparticles:
governing
physical
parameters.
Molecular
Pharmaceutics, v. 26, n. 1, p. 1–7, 2009.
LACHMAN, L.; LIEBERMAN, H.; KANIG, J. Teoria e prática na indústria
farmacêutica. 2001.
LAI, F.; SINICO, C.; ENNAS, G. Diclofenac nanosuspensions: influence of
preparation procedure and crystal form on drug dissolution behaviour. International
journal of pharmaceutics, v. 373, n. 1-2, p. 124–32, 21 maio 2009.
LE, Y. et al. Nanosized bicalutamide and its molecular structure in solvents.
International journal of pharmaceutics, v. 370, n. 1-2, p. 175–80, 31 mar. 2009.
LI, X. et al. Preparation of uniform prednisolone microcrystals by a controlled
microprecipitation method. International journal of pharmaceutics, v. 342, n. 1-2,
p. 26–32, 5 set. 2007.
MANADAS, R.; PINA, M.; VEIGA, F. A dissolução in vitro na previsão da absorção
oral de fármacos em formas farmacêuticas de liberação modificada. Revista
Brasileira de Ciências Farmacêuticas, v. 38, 2002.
MASTERTON, W. L.; SLOWINSKI, E. J.; STANITSKI, C. L. Princípios de química.
[s.l.] Guanabara Koogan, 1990.
MATTEUCCI, M. et al. Drug nanoparticles by antisolvent precipitation: mixing energy
versus surfactant stabilization. Langmuir, v. 22, n. 21, p. 8951–9, 10 out. 2006.
MENG, X.; CHEN, Y.; CHOWDHURY, S. Stabilizing dispersions of hydrophobic drug
molecules using cellulose ethers during anti-solvent synthesis of micro-particulates.
Colloids and Surfaces B: Biointerfaces, v. 70, p. 7–14, 2009.
MINÉ, T.; MORAIS, D. DE. Revisão Das Legislações Que Vigoram Sobre A
Estabilidade Dos Medicamentos Na Indústria Farmacêutica Brasileira. FOCO, p. 21–
38, 2013.
MISRA, S. K. et al. The complexity of nanoparticle dissolution and its importance in
nanotoxicological studies. The Science of the total environment, v. 438, p. 225–32,
1 nov. 2012.
MOTA, F. L. et al. Solubility of drug-like molecules in pure organic solvents with the
CPA EoS. Fluid Phase Equilibria, v. 303, n. 1, p. 62–70, abr. 2011.
MOU, D. et al. Potent dried drug nanosuspensions for oral bioavailability
enhancement of poorly soluble drugs with pH-dependent solubility. International
journal of pharmaceutics, v. 413, n. 1-2, p. 237–44, 15 jul. 2011.
MUDIE, D. M.; AMIDON, G. L.; AMIDON, G. E. Physiological parameters for oral
delivery and in vitro testing. Molecular Pharmaceutics, v. 7, p. 1388–1405, 2010.
75
PAUL, D. R. Elaborations on the Higuchi model for drug delivery. International
journal of pharmaceutics, v. 418, n. 1, p. 13–7, 10 out. 2011.
PHARMACOPOEIA, B. The British Pharmacopoeia Secretariat. Market Towers,
2009.
PORTUGAL. Farmacopeia Portuguesa VII. 7°. ed. Lisboa: Ministério da Saúde:
Infarmed, 2002.
POUTON, C. W. Formulation of poorly water-soluble drugs for oral administration:
physicochemical and physiological issues and the lipid formulation classification
system. European journal of pharmaceutical sciences, v. 29, n. 3-4, p. 278–87,
nov. 2006.
RASENACK, N.; HARTENHAUER, H.; MÜLLER, B. W. Microcrystals for dissolution
rate enhancement of poorly water-soluble drugs. International Journal of
Pharmaceutics, v. 254, n. 2, p. 137–145, mar. 2003.
RAW, A. S. et al. Regulatory considerations of pharmaceutical solid polymorphism in
Abbreviated New Drug Applications (ANDAs). Advanced drug delivery reviews, v.
56, n. 3, p. 397–414, 23 fev. 2004.
RODRIGUES, P.; SILVA, M. Avaliação in vitro de medicamentos de liberação
prolongada: aplicação de métodos estatísticos, modelos dependentes e
independentes de análise. Revista colombiana de ciencias quimicofarmaceuticas, 2005.
ROSSMANN, M. et al. Solute solubility as criterion for the appearance of amorphous
particle precipitation or crystallization in the supercritical antisolvent (SAS) process.
The Journal of Supercritical Fluids, v. 66, p. 350–358, jun. 2012.
ROWE, R.; SHESKEY, P. Handbook of pharmaceutical excipients. 6°. ed. [s.l.]
Pahmaceutical Press, 2006.
SCHESHOWITSCH, K.; PEREIRA, A. Avaliação da qualidade e perfil de dissolução
de cápsulas manipuladas de piroxicam. Latin American Journal of Phamacy, v. 26,
n. 5, p. 645–651, 2007.
SERRA, C.; STORPIRTIS, S. Comparação de perfis de dissolução da cefalexina
através de estudos de cinética e eficiência de dissolução (ED por cento). Revista
Brasileira de Ciências Farmacêuticas, v. 43, 2007.
SHARGEL, L.; YU, A. Applied biopharmaceutics and pharmacokinetics. 4 ed ed.
Stamfor: Appleton & Lange, 1999. p. 768
SIEPMANN, J.; SIEPMANN, F. Mathematical modeling of drug dissolution.
International journal of pharmaceutics, v. 453, n. 1, p. 12–24, 30 ago. 2013.
76
SILVERSTEIN, R. M. .; WEBSTER, F. X. .; KIEMLE, D. J. Identificação
Espectrométrica de Compostos Orgânicos. 7°. ed. Rio de Janeiro: LTC - Livros
Técnicos e Científicos Editos S.A, 2006.
SINGHVI,
G.;
SINGH,
M.
REVIEW :
IN-VITRO
DRUG
RELEASE
CHARACTERIZATION MODELS. International journal of pharmaceutical Studies
and Research, v. II, n. I, 2011.
STEWART, P. J.; ZHAO, F.-Y. Understanding agglomeration of indomethacin during
the dissolution of micronised indomethacin mixtures through dissolution and deagglomeration modeling approaches. European journal of pharmaceutics and
biopharmaceutics, v. 59, n. 2, p. 315–23, fev. 2005.
STORPIRTIS, S.; MARCOLONGO, R. A equivalência farmacêutica no contexto da
intercambialidade entre medicamentos genéricos e de referência: bases técnicas e
científicas. Infarma, v. 16, n. 9-10, p. 51–56, 2004.
TAUBØLL, E. et al. A new injectable carbamazepine solution — antiepileptic effects
and pharmaceutical properties. Epilepsy Research, v. 7, n. 1, p. 59–64, set. 1990.
THIERING, R.; DEHGHANI, F.; FOSTER, N. R. Current issues relating to antisolvent micronisation techniques and their extension to industrial scales. The
Journal of Supercritical Fluids, v. 21, n. 2, p. 159–177, out. 2001.
THORAT, A. A.; DALVI, S. V. Liquid antisolvent precipitation and stabilization of
nanoparticles of poorly water soluble drugs in aqueous suspensions: Recent
developments and future perspective. Chemical Engineering Journal, v. 181-182,
p. 1–34, fev. 2012.
USP. United States Pharmacopeial Convention. [s.l.] United States Pharmacopeial
Convention, 2006.
VERMA, S.; GOKHALE, R.; BURGESS, D. J. A comparative study of top-down and
bottom-up approaches for the preparation of micro/nanosuspensions. International
journal of pharmaceutics, v. 380, n. 1-2, p. 216–22, 1 out. 2009.
VIÇOSA, A. et al. An innovative antisolvent precipitation process as a promising
technique to prepare ultrafine rifampicin particles. Journal of Crystal Growth, v.
342, n. 1, p. 80–87, mar. 2012.
VIEHOF, A. et al. Oral insulin delivery in rats by nanoparticles prepared with nontoxic solvents. International journal of pharmaceutics, v. 443, n. 1-2, p. 169–74, 25
fev. 2013.
WANG, Z.; CHEN, J.; LE, Y. Preparation of ultrafine beclomethasone dipropionate
drug powder by antisolvent precipitation. Industrial & Engineering Chemistry
Research, p. 4839–4845, 2007.
WONG, S. M.; KELLAWAY, I. W.; MURDAN, S. Enhancement of the dissolution rate
and oral absorption of a poorly water soluble drug by formation of surfactant-
77
containing microparticles. International journal of pharmaceutics, v. 317, n. 1, p.
61–8, 6 jul. 2006.
XIE, Y. et al. Quantitative determination of solid-state forms of a pharmaceutical
development compound in drug substance and tablets. International journal of
pharmaceutics, 2008.
YOUN, Y. et al. Dissolution rate improvement of valsartan by low temperature
recrystallization in compressed CO 2 : Prevention of excessive agglomeration. The
Journal of Supercritical Fluids, v. 59, p. 117–123, 2011.
ZHANG, G. G. Z. et al. Phase transformation considerations during process
development and manufacture of solid oral dosage forms. Advanced drug delivery
reviews, v. 56, n. 3, p. 371–90, 23 fev. 2004.
ZHANG, H. X. et al. Micronization of atorvastatin calcium by antisolvent precipitation
process. International Journal of Pharmaceutics, v. 374, p. 106–113, 2009.
ZHANG, J.-Y. et al. Preparation of amorphous cefuroxime axetil nanoparticles by
controlled nanoprecipitation method without surfactants. International journal of
pharmaceutics, v. 323, n. 1-2, p. 153–60, 12 out. 2006.
ZHAO, H. et al. Controlled Liquid Antisolvent Precipitation of Hydrophobic
Pharmaceutical Nanoparticles in a Microchannel ReZHAO, H.; WANG, J.-X.; WANG,
Q.-A.; CHEN, J.-F.; YUN, J. Controlled Liquid Antisolvent Precipitation of
Hydrophobic Pharmaceutical Nanoparticles in. Industrial & Engineering Chemistry
Research, v. 46, n. 24, p. 8229–8235, nov. 2007.