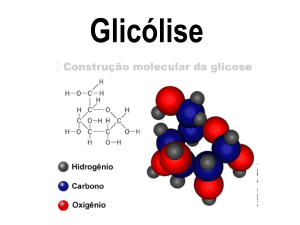
2.4- Oxidação metabólica da glicose
A glicose ocupa uma posição central no metabolismo de muitos seres vivos,
apresentando um nível relativamente alto de energia potencial, o que a torna um bom
combustível para as reacções que ocorrem no ambiente intracelular. Este facto é potenciado
pela possibilidade de armazenamento celular em formas poliméricas de elevado peso
molecular (amido, glicogénio, etc.) que são compatíveis homeostaticamente (não desregulam
os níveis de glicose no sangue). A glicose, em situações de exigência energética, vai ser
libertada destas formas poliméricas de armazenamento, ficando disponível para entrar em
processos de oxidação e consequente extracção de ATP. É de realçar, que a glicose é também
usada como percursor de inúmeros intermediários metabólicos em reacções de biossíntese.
De uma forma geral, podemos apontar três vias metabólicas principais para a glicose:
o seu armazenamento (como polissacárido);
a sua oxidação pela via das pentoses-fosfato, originando ribose-5-fosfato para a síntese de
ácidos nucleicos, e de NADPH para processos de redução;
a oxidação via glicólise originando piruvato e providenciando ATP e intermediários
metabólicos de outras vias.
A glicólise consiste numa série de dez reacções químicas, catalisadas por enzimas, nas
quais uma molécula de glicose vai ser degradada em duas moléculas de piruvato. Durante a
sequência de reacções, uma parte da energia livre, proveniente da degradação da glicose, vai
ser conservada em ATP e NADH. Este processo é o caminho central no catabolismo da
glicose e é de uma importância vital para inúmeros organismos, alguns dos quais têm neste
processo, a sua única fonte de energia metabólica.
É comum dividir a glicólise em duas fases distintas: a fase de preparação e a fase de oxiredução; a cada uma delas vão corresponder cinco reacções químicas.
Na primeira fase gastam-se 2 moléculas de ATP em duas fosforilações; esta fase acaba
com a formação de 2 trioses, 2 moléculas de gliceraldeído 3-fosfato, que resultam da
clivagem da glicose.
Na fase de oxi-redução vai haver um retorno do investimento de 2 moléculas de ATP da
fase anterior: vão ser formadas 4 moléculas de ATP (através da fosforilação de ADP), para
além de 2 moléculas de NADH, por cada molécula de glicose. Esta fase termina com a
formação de piruvato.
1
A equação geral da glicólise é então a seguinte:
Esta equação pode ser separada em 2 processos distintos: por um lado a conversão de
glicose a piruvato, uma reacção exergónica, e por outro a formação de ATP a partir de ADP
e Pi, endergónica. Na soma das duas, conclui-se que a glicólise tem uma variação de energia
livre padrão de -85kJ/mol.
O processo de glicólise está esquematizado na figura seguinte:
Sequência Reacções Glicólise
2
1. A molécula de glicose é fosforilada no carbono 6’, por acção da hexoquinase, formando
glucose-6-fosfato, Há gasto de uma molécula de ATP.
2. A glicose-6-fosfato vai sofrer uma isomerização, por acção da fosfohexose isomerase,
formando-se então frutose-6-fosfato.
3. Fosforilação da frutose-6-fosfato no carbono 1, mais uma vez com gasto de um ATP,
formando-se frutose-1,6 bifosfato. Esta reacção é o primeiro e mais importante ponto
de regulação da glicólise: a formação de frutose 1,6-bifosfato é exclusiva da via
metabólica da glicólise. Além disso, a enzima PFK-1 é uma enzima reguladora cuja
actividade é aumentada quando a célula entra em necessidades energéticas (relação
[ATP]/[ADP] menor que 1).
4. Clivagem da frutose 1,6-bifosfato que origina 2 trioses fosfatadas: o gliceraldeído 3fosfato e a dihidroxiacetona fosfato.
5. Apenas uma destas trioses pode ser directamente degradada nos processos seguintes da
glicólise, que é o gliceraldeído 3- fosfato. Há então a conversão rápida da
dihidroxiacetona fosfato em gliceraldeído 3-fosfato pela triose-fosfato isomerase. Chegase ao final da fase preparatória e a molécula de glicose inicial dá origem a 2 moléculas de
gliceraldeído 3-fosfato.
6. O gliceraldeído formado na fase preparatória vai ser oxidado a 1,3-bifosfoglicerato, não
para um grupo carboxilo livre, mas com a ajuda do fosfato inorgânico. O aceitador de
hidrogénios é o NAD+, formando-se NADH + H+.
7. A enzima fosfoglicerato quinase catalisa a transferência do fosforilo do grupo carboxilo
do 1,3-bifosfoglicerato para um ADP, formam-se 2 moléculas de ATP e de 3fosfoglicerato. Esta reacção e a anterior juntas constituem um processo conjunto em que o
1,3-bifosfoglicerato é o produto intermédio: no total das 2 reacções, a reacção de
formação de ATP acaba por ser exergónica – tipo de formação de ATP dita como
fosforilação ao nível do substrato (intervém o o 1,3-bifosfoglicerato).
8. Troca entre o grupo fosforilo e o hidroxilo do glicerato, formando-se então 2fosfoglicerato, num processo que ocorre em 2 etapas com a ajuda a fosfoglicerato mutase.
9. Nesta reacção, dá-se a desidratação do 2-fosfoglicerato a fosfoenolpiruvato (PEP), por
acção da enolase: há a remoção de 1 molécula de água.
10. No último passo da glicólise, que é também um importante ponto regulador de todo o
processo, a piruvato quinase desfosforila o fosfoenolpiruvato para o ADP, formando-se
mais uma vez 2 moléculas de ATP e o produto final da glicólise, o piruvato (num
primeiro momento é formada a forma enol do piruvato, o enolpiruvato e só depois adquire
a forma ceto - simplesmente piruvato).
3
Analisando o destino dos produtos finais da glicólise, em condições aeróbias, a glicólise
é apenas a primeira etapa da degradação completa da glicose:
as 2 moléculas de NADH vão ser reoxidadas na cadeia respiratória, na mitocôndria, o
que fornece a energia para a síntese de ATP por fosforilação oxidativa;
O piruvato vai ser oxidado e transformado em acetato, que constituirá o grupo acetilo
da acetil-CoA. Esta segue para o ciclo de Krebs, sendo oxidada a CO2;
No total, 30 a 32 moléculas de ATP são formadas por cada molécula de glicose.
Em condições anaeróbias, o piruvato vai entrar em processos de fermentação láctica ou
alcoólica.
No caso da fermentação láctica, dá-se a redução do piruvato a lactato: o piruvato
aceita os electrões do NADH e regenera NAD+, que permite a continuação da glicólise;
Na fermentação alcoólica, temos a conversão do piruvato em etanol e C02 através de
2 passos. No primeiro passo, há uma descarboxilação irreversível do piruvato formandose acetaldeído. No 2º passo o acetaldeído é reduzido a etanol, através da acção do álcool
desidrogenase e do poder redutor do NADH, formando-se ainda CO2;
Formam-se 2 moléculas de ATP.
A regulação do mecanismo de acção da glicólise é conseguida através de um complexa
interacção entre os níveis [ATP]/[ADP] presentes na célula, regeneração de NADH e
regulação alostérica de várias enzimas, nomeadamente a hexoquinase, PFK-1 e a piruvato
quinase (G bastante negativos em todas as reacções catalisadas por estas enzimas). Para
além disso, existem certos metabolitos que dão a informação e a percepção se a célula está
com energia em excesso ou se pelo contrário, necessita de uma activação do mecanismo da
glicólise para se obter mais ATP.
O piruvato, em condições aeróbias, pode ser oxidado originando o grupo acetil da
acetil-CoA (libertando-se dióxido de carbono) que irá posteriormente entrar no ciclo de
Krebs. Esta descarboxilação oxidativa é catalisada pelo complexo piruvato-desidrogenase
(PDH). Este complexo é constituído por múltiplas cópias de três enzimas distintas - E1
(piruvato
desidrogenase),
E2
(dihidrolipoilo
transacetilase)
e
E3
(dihidrolipoilo
desidrogenase) - e necessita de 5 coenzimas: tiamina pirofosfato (TPP), flavina adenina
dinucleótido (FAD), Coenzima A (CoA), nicotinamida adenina dinucleótido (NAD) e
Lipoato. São também componentes vitais deste sistema em cada um destes grupos prostéticos
as vitaminas tiamina (no TPP), riboflavina (no FAD), niacina (no NAD) e pantotenato (na
CoA).
4
A coenzima A é formada por pantotenato, 3-fosfoadenosina difosfato (forma
fosforilada de ADP) e β-mercato-etilamina qu possui um grupo tiol (-SH) muito reactivo que
é fundamental no papel da CoA como transportadora de grupos acilo, como por exemplo o
acetilo, que forma a acetil-coA. Os grupos acetil ligam-se a este grupo tiol formando
tioésteres; já o o lipoato possui dois grupos tiol podendo formar ligações dissulfito através de
oxidação, sendo um bom transportador de H+ e de grupos acetil simultaneamente.
Analisando mais pormenorizadamente a estrutura do PDH vemos que a enzima E2 tem
três subunidades/domínios distintos, um é o local de ligação do lipoato ao resíduo de Lis desta
enzima, outro é o local de ligação de E1 e E3 e o terceiro é o centro activo, acetiltransferase.
O TPP liga-se ao centro activo de E1 e o FAD ao centro activo de E3.
A acção do complexo PDH é um exemplo de canalização do substrato, a acção da
enzima sobre o substrato dá-se por “arrasto”, não havendo libertação de produtos
intermediários para fora do complexo, reagindo assim mais rapidamente.
No primeiro passo, dá-se a descarboxilação do piruvato pelo TPP da enzima E1, formandose hidroxietilo TPP, sendo nesta reacção que o complexo PDH exerce a sua função de
especificidade de substrato. No passo 2, há oxidação do grupo hidroxietilo a acetato que se
liga a um dos grupos tiol do lipoato, ficando o outro reduzido a SH. O lipoato transporta
então o acetato até à coenzima A, ligando-se este ao grupo tiol desta coenzima. Nos passos
4 e 5 há reoxidação dos grupos tiol do lipoato para este iniciar nova ronda de oxidação do
piruvato. Esta reoxidação dos grupos tiol dá-se por redução do FAD (presente da E3) e
posteriormente do NAD+.
A regulação da produção de acetil-CoA é feita pelo produto, ou seja ATP, acetil-CoA,
NADH e mesmo ácidos gordos de cadeia longa inibem o complexo PDH. Pelo contrário,
AMP, CoA e NAD+ activam o complexo, dado que a sua maior concentração indica um
fluxo menor de produção de acetil-CoA. De um modo geral, a reacção de oxidação do
piruvato é activada em situações de maior exigência energética, encaminhando mais acetilCoA para o Ciclo de Krebs.
5
Em mamíferos, esta regulação é ainda complementada por modificação covalente da
estrutura proteica. O complexo PDH é inibido por fosforilação de um resíduo de Ser numa
das subunidades de E1 por uma proteína cinase. A E1 é activada novamente pela acção da
outra proteína reguladora existente no complexo, uma fosfoproteina que vai remover o grupo
fosforilo por hidrólise, tendo portanto acção contrária à cinase. A acção destas proteínas é
regulada pela relação [ATP]/[ADP]. Uma maior relação [ATP]/[ADP] activa a proteína
cinase inibindo a produção de acetil-CoA e uma menor relação [ATP]/[ADP] activa a
fosfoproteína activando novamente o complexo PDH.
6
2.5- Regulação da Glicólise, Oxidação de Hexoses,
Via Das Fosfopentotoses
Mecanismos de regulação não hormonal da glicólise
As oses, em particular a glicose, devem a sua importância ao facto de a sua oxidação
fornecer aos organismos vivos grande parte da energia que lhes é necessária.
A glicólise processa-se no citosol e é regulada por três enzimas: a primeira regula a
entrada de glicose na via glicolítica, já que é a enzima que catalisa a primeira reacção desta
via metabólica, e as outras duas regulam a via propriamente dita.
É importante frisar que as três enzimas reguladoras correspondem às enzimas que
catalisam reacções unidireccionais, isto é, as enzimas que catalisam as reacções inversas
(gliconeogénese) não são as mesmas que catalisam as reacções na glicólise.
Regulação da entrada de glicose na via glicolítica - Hexocinase:
Esta enzima catalisa a fosforilação da glicose em glicose-6-fosfato.
As hexocinases I,II e III são inibidas pelo produto da reacção, glicose-6-fosfato. Se a
metabolização da glicose-6-fosfato é menor que a sua síntese, esta acumula-se inibindo a
hexocinase.
Por outro lado, a hexocinase IV, predominante no fígado, não sofre regulação alostérica
pela Glicose-6P, e possui um valor de Km mais elevado, pelo que a sua saturação ocorre para
níveis mais elevados de glicose, permitindo a sua eficácia no metabolismo de altos níveis de
glicose. A sua regulação consiste na inibição por uma proteína específica, que pode ser
dissociada pela molécula de glicose (ficando a enzima activa). Caso haja uma baixa
concentração de glicose no sangue, ocorre inactivação da hexocinase IV pela frutose-6P
(através do transporte da enzima para o nécleo da célula onde se liga reversivelmente à
proteína reguladora); uma vez restaurados os níveis de glicose, esta reverte o processo
anterior, dissociando a proteína reguladora da hexocinase IV, que readquire a sua capacidade
catalítica.
Regulação da via propriamente dita:
Fosfofrutocinase-1:
A fosfofrutocinase-1 é um enzima muito complexa que catalisa a fosforilação da frutose-6fosfato em frutose-1,6-bisfosfato - terceira etapa da via glicolítica.
A regulação alostérica desta enzima é coordenada por vários activadores e inibidores.
7
Em relação aos activadores: AMP, ADP (indicadores de falta de moléculas energéticas –
ATP), e a frutose-2,6-bisfosfato (que não é uma composto intermediário da glicólise, e pode
ser indicadora de uma falha em reacções de fosforilação, induzindo a produção de frutose-1,6bisP, que já é um intermediário da glicólise e possibilita a sua continuação) .
Quanto aos inibidores: ATP (a sua abundância relativa indica disponibilidade de energia),
frutose-1,6-bisfosfato e citrato (que indica a abundância de intermediários do ciclo de Krebs).
Piruvato-cinase:
A piruvato-cinase catalisa a última reacção da via, a conversão de fosfoenolpiruvato em
piruvato.
A sua regulação alostérica consiste no activador frutose 1-6-bisfosfato, e numa série de
inibidores: ATP, Acetil-CoA, ácidos gordos de cadeia longa (indicadores da presença de
fontes de energia) e a alanina (que pode ser sintetizada a partir do piruvato por
transaminação).
Existem duas isoenzimas principais de piruvato-cinase: L (fígado – liver), que possui
regulação hormonal, e M (músculo).
Oxidação de outras Hexoses
Frutose:
A frutose pode ser obtida, na sua forma livre, pela ingestão de frutas, ou como
constuituínte do dissacárido sacarose, a qual é hidrolisada pela enzima sacarase (obtendo-se
uma molécula de frutose e outra de glicose). É incorporada na via glicolítica de duas formas
distintas, dependendo do local onde o seu metabolismo decorre.
O primeiro passo será a fosforilação da frutose, pela enzima hexocinase:
Frutose + ATP
Mg2+
Frutose 6-fosfato + ADP
sendo o produto frutose 6-fosfato incorporado na via glicolítica (transformada em frutose 1,6bisfosfato, continuando a via metabólica).
Por outro lado, se a fosforilação da frutose ocorrer no fígado, onde se encontra presente a
enzima frutocinase, a fosforilação ocorre no carbono C1 da frutose, ao invés do carbono C6:
Frutose + ATP
Mg2+
Frutose 1-fosfato + ADP
A frutose 1-fosfato sofre clivagem em gliceraldeído e di-hidroxiacetona-fosfato pela
enzima frutose 1-fosfato aldolase:
Frutose 1-fosfato
gliceraldeído + di-hidroxiacetona-fosfato
A di-hidroxiacetona-fosfato é transformada em gliceraldeído 3-fosfato pela triose-fosfato
isomerase, e o gliceraldeído é fosforilado pela enzima triose cinase, formando gliceraldeído 3fosfato.
As duas moléculas de gliceraldeído 3-fosfato obtidas são incorporadas na glicólise.
8
Galactose:
A galactose é obtida por hidrólise da lactose (açúcar do leite), catalisada pela enzima
lactase, da qual resulta uma molécula de galactose e uma de glicose.
A galactose começa por ser fosforilada, no fígado, pela enzima galactocinase:
Galactose + ATP
Mg2+
Galactose-1-fosfato + ADP
A conversão da galactose 1-fosfato no epímero glicose-1-fosfato é realizada na sua forma
conjugada com o transportador uridina difosfato (UDP), que actua como coenzima de
transferência de grupos.
(1) A galactose-1-fosfato é transformada em UDP-galactose, pela enzima galactose 1fosfato uridiltransferase – ocorre transferência do grupo UDP-glicose para a galactose-1-P.
(2) A UDP-galactose é transformada em UDP glicose pela enzima UDP-glicose 4epimerase, ocorrendo a oxidação do grupo OH de C4, no qual resulta um grupo cetona, e
posterior redução deste em hidroxilo, com inversão da orientação (epimerização). O NAD é o
cofactor para a oxidação e para a redução.
(3) A UDP-glicose intervém na reacção (1), fornecendo o seu grupo UDP à galatose 1fosfato, e havendo libertação de glicose 1-fosfato.
A glicose 1-fosfato obtida é transformada em glicose-6-fosfato pela enzima
fosfoglicomutase, sendo a G6P incorporada na glicólise.
Manose:
A manose é ingerida na forma de polissacáridos ou de glicopoteínas. O seu metabolismo
consiste na sua fosforilação pela enzima hexocinase:
Manose + ATP
Mg2+
Manose 6-fosfato + ADP
A enzima fosfomanose isomerase será a responsável pela transformação da manose 6fosfato em frutose 6-fosfato, que pode ser degrada pela via glicolítica.
A oxidação de monossacáridos como a frutose, galactose e manose permite a obtenção de
energia a partir de moléculas alternativas à glicose, o que se revela extremamente vantajoso
em situação de défice de glicose. Assim como a glicose, a degradação da frutose, galactose e
manose requer o investimento de moléculas de ATP na fosforilação destas moléculas,
investimento esse que será compensado posteriormente, ocorrendo formação de moléculas de
ATP e de moléculas com poder redutor (NADH + H+), que poderão também ser utilizadas
para obtenção de energia na cadeia respiratória.
É de salientar que o metabolismo das oses descrito possui uma regulação parcialmente
diferente da glicólise, uma vez que os compostos que são incorporados na glicólise não foram
alvo do primeiro passo de regulação da glicólise: regulação pela enzima hexocinase. Tal facto
pode explicar a menor velocidade de metabolização da glicose face à das outras oses.
9
Via das Fosfopentoses
Na maioria das células, o maior destino catabólico da Glicose-6-Fosfato é a glicólise. No
entanto, cerca de 10% dessa molécula será também degradado pela Via das Fosfopentoses.
Esta via metabólica ocorre em células de divisão rápida (como a medula óssea, a pele ou a
mucosa intestinal), em tecidos com extensa síntese de ácidos gordos (como o fígado, tecido
adiposo e glândulas mamárias em lactação), ou síntese activa de hormonas esteróides (como
as gónadas).
As principais funções desta via metabólica são: a formação de DNA e RNA, a síntese de
coenzimas como o ATP, NADH, FADH2 e Coenzima A.
Todas as enzimas intervenientes na via das fosfopentoses encontram-se no citosol celular.
1ª Fase – Fase das reacções oxidativas e irreversíveis ou fase das desidrogenases
Glicose-6-Fosfato
NADP+
Enzima: Glicose-6-P Desidrogenase.
O NADP+ é reduzido.
É essencial a presença de Mg2+ ou Ca2+ para se dar
esta reacção
NADPH
6-Fosfogliconolactona
H2O
Enzima: 6-fosfogliconolactonase (hidrolase)
Apesar de esta reacção ser espontânea (exergónica), é utilizada
esta enzima para garantir a total conversão das moléculas
reagentes nos produtos.
É libertada uma molécula de água (reacção de hidrólise).
É essencial a presença de Mg2+, Mn2+ ou Ca2+ para se dar esta
reacção.
6-Fosfogliconato
NADP+
CO2
NADPH
Enzima: 6-Fosfogliconato Desidrogenase
Reacção de oxidação e descarboxilação.
Redução do NADP+
É essencial a presença de Mg2+, Mn2+ ou Ca2+ para
se dar esta reacção.
Ribulose-5-Fosfato
Enzima: pentose-fostato isomerase
Reacção de isomerização.
Ribose-5-fosfato
10
2ª Fase – Fase das reacções reversíveis não oxidativas
É importante referir que a reacção de isomerização da ribulose-5-P em ribose-5-P ainda faz
parte da primeira fase desta via metabólica.
Regulação da Via Metabólica
A regulação desta via metabólica é efectuada tanto a nível da síntese das enzimas que
catalisam as reacções da via, como de regulação alostérica por parte dos substratos e
metabolitos.
Regulação por síntese de enzimas:
A regulação por síntese de enzimas consiste na regulação da transcrição dos genes que
codificam as enzimas catalíticas desta via, aumentando ou diminuindo a sua produção em
função das necessidades da célula.
Desidrogenases: síntese é induzida por hormonas (insulina e tiroxina)
Transcetolase: síntese controlada pela Vitamina B1 na sua forma activa, Tiamina
Pirofosfato.
Regulação alostérica:
A regulação alostérica consiste na inibição da enzima glicose-6-fosfato desidrogenase por
parte do NADPH. Dado que a glicose-6-P existente na célula é direccionada para a glicólise
ou para a via das fosfopentoses de acordo com as necessidades da célula, caso esta necessite
11
(ou não) de produzir moléculas redutoras de NADPH. Assim sendo, faz sentido que esta
molécula seja um regulador alostérico da enzima que catalisa a entrada da glicose-6-fosfato na
via das fosfopentoses (G6P desidrogenase). Para altos níveis de NADPH, ocorre inibição da
enzima G6P desidrogenase, e consequente inbição da própria via das fosfopentoses; a glicose6-fosfato segue, portanto, a via glicolítica. Por outro lado, quando existe baixa concentração
de NADP+ no citosol, existe necessidade de produção de moléculas de NADPH, pelo que a
enzima G6P desidrogenase irá ser estimulada alostericamente pelo NADP+ e irá encaminhar a
G6P para a via das fosfopentoses.
Destino Metabólico dos Intervenientes
Com a realização da via das fosfopentoses, são formados compostos que possuem diversas
funcionalidades na célula.
O produto final da primeira fase desta via metabólica, a ribose-5-fosfato, é um percursor na
síntese de nucleótidos para incorporação destes nos ácidos nucleicos.
O NADPH formado na fase oxidativa possui um papel fulcral na manutenção de um
ambiente redutor no interior da célula: a sua oxidação possibilita a redução do glutatião, que
na sua forma reduzida, constitui uma eficiente protecção das proteínas, lípidos e outros
compostos de elevada importância biológica contra as moléculas ou radicais oxidantes que
possam existir na célula, como o radical superóxido e o hidroxilo, ou a molécula de peróxido
de hidrogénio. Como exemplo desta função são os eritrócitos, nos quais existem grandes
quantidades de oxigénio molecular, que é um forte oxidante, havendo a necessidade constante
de prevenir e reverter oxidações de biomoléculas.
O NADPH constitui ainda um elemento fundamental nas reacções de biossíntese redutora
de ácidos gordos e outras moléculas, actuando como agente redutor.
Na segunda fase da via das fosfopentoses, há formação de compostos intermediários da via
glicolítica: frutose-6-fosfato e gliceraldeído-3-fosfato. Estes compostos podem incorporar-se
na via glicolítica, quer no sentido da realização da glicólise, quer no sentido da
gliconeogénese, no qual há formação de glicose-6-fosfato que poderá reintegrar novamente a
via das fosfopentoses.
12
2.6- Catabolismo de Ácidos Gordos e Cetogénese
Uma vez que já foram previamente abordados os conceitos básicos relativos aos lípidos,
assim como a sua digestão e transporte, vamos então focar a oxidação dos ácidos gordos, um
conjunto de três vias que tem como objectivo final a obtenção de energia. De facto, o
catabolismo de ácidos gordos é uma das fontes de energia mais importantes para muitos
organismos, estando responsável, por exemplo, por cerca de 80% das necessidades
energéticas do coração e fígado dos mamíferos.
Em primeiro lugar temos a β-oxidação, na qual os ácidos gordos sofrem
sucessivamente a remoção de pares de carbonos sob a forma de acetil-CoA, com início no
terminal carboxil da cadeia. Se pensarmos no caso do ácido palmítico, que possui dezasseis
carbonos, são necessárias sete passagens pela cadeia oxidativa para que restem apenas dois
carbonos que são automaticamente convertidos em acetil-CoA; logo, obtém-se um total de
oito unidades de acetil-CoA. Note-se que a formação de cada molécula de acetil-CoA implica
a remoção de quatro átomos de hidrogénio (4 H+ e 4 e-).
Na segunda fase apresenta-se o ciclo do ácido cítrico, em que o facto mais importante,
neste caso, é o de as moléculas de acetil-CoA provenientes da β-oxidação serem oxidadas a
CO2, juntando-se às moléculas derivadas da glicose (via glicólise e oxidação do piruvato).
Estas duas fases têm lugar na mitocôndria e reduzem os transportadores de electrões
NADH e FADH2.
Por último, temos na terceira fase a cadeia respiratória, na qual os electrões têm como
aceitador final o O2, dando-se a formação de H2O e ATP.
β-Oxidação de Ácidos Gordos Saturados com Número Par de Carbonos
Embora a β-oxidação se altere naturalmente de organismo para organismo, o essencial
do processo mantém-se: é composta por quatro passos repetidos tantas vezes quantas
necessárias. Trata-se de uma espécie de maneira elegante de quebrar uma ligação –CH2-,
relativamente estável por natureza. A preparação nos três primeiros passos de uma ligação CC menos forte, com uma cetona, faz com que esta seja um alvo mais fácil para um ataque
nucleofílico pelo grupo –SH da Coenzima A no passo final.
No primeiro passo da β-oxidação, observa-se a desidrogenação do grupo acil-CoA, o
que vai induzir a formação de uma ligação dupla entre os Carbonos α e β, resultando num
novo composto designado trans-Δ2-Enol-CoA (o Δ2 designa a posição da ligação dupla). Este
novo composto tem a configuração trans, embora normalmente as ligações nos ácidos gordos
insaturados sejam cis.
13
Esta reacção é catalisada por três isoenzimas da acil-CoA desidrogenase, cuja utilização
depende do tamanho da cadeia: a “very-long-chain acyl-CoA dehydrogenase” (VLCAD), que
actua em ácidos gordos que tenham entre doze e dezoito carbonos; a “medium-chain”
(MCAD), para cadeias com quatro a catorze carbonos e a “short-chain” (SCAD), no caso de
cadeias com quatro a oito carbonos. Estas isoenzimas são flavoproteínas que possuem o FAD
como grupo prostético. Uma vez este reduzido, irá doar imediatamente os electrões para a
cadeia respiratória, com o auxílio da “electron transferring flavoprotein” (ETF).
Neste passo junta-se água à ligação dupla entre os carbonos α e β, com o objectivo
de formar o estereoisómero L do β-hidroxiacil-CoA, o 3-hidroxiacil-CoA. Esta reacção é
catalisada
pela
enol-CoA hidratase, sendo análoga à reacção da fumarase no ciclo do ácido cítrico.
No terceiro passo dá-se a desidrogenação do L-β-hidroxiacil-CoA a β-cetoacil-CoA.
A enzima interveniente é a β-hidroxiacil-CoA desidrogenase, sendo o aceitador de electrões o
NAD+. Esta reacção é análoga à que se dá na desidrogenação do malato no ciclo do ácido
cítrico.
O quarto e último passo é catalisado por uma enzima que se chama vulgarmente tiolase,
mas cuja designação correcta é acil-CoA acetiltransferase. A função da tiolase é promover a
ligação do cetoacil-CoA a uma molécula livre de Coenzima A, o que quebrará a ligação com
o terminal carboxil, libertando-se um acetil-CoA e ficando o ácido gordo dois carbonos mais
curto. Pode fazer-se uma analogia entre esta reacção e a hidrólise, uma vez que o β-cetoacilCoA é clivado pelo grupo tiol da coenzima A.
Os três últimos passos da β-Oxidação são catalisados por um conjunto diferente de
enzimas conforme o tamanho do ácido gordo em questão: no caso de a cadeia ter mais de
doze carbonos, é usada a proteína trifuncional (TFP), que consiste num agregado de três
proteínas muito eficiente na sua função devido à proximidade entre os centros activos das
proteínas que o compõem; para cadeias com até doze carbonos, existe um conjunto de quatro
enzimas solúveis na matriz mitocondrial.
Sendo a β-oxidação a primeira parte do catabolismo dos ácidos gordos, esta é
responsável pela produção de três tipos de produtos: acetil-CoA (um por cada passagem),
electrões (dois pares por passagem, nos transportadores adequados, NADH e FADH2) e
catiões H+ (quatro por passagem). O acetil-CoA pode ser oxidado a CO2 e H2O no ciclo do
ácido cítrico, que irá também implicar a libertação de electrões. Os H+ e os electrões
provenientes tanto da β-oxidação como do ciclo do ácido cítrico prosseguem então para a
cadeia respiratória, num processo que já foi descrito em apresentações anteriores, cujos
produtos são água e energia sob a forma de ATP.
14
Catabolismo dos ácidos gordos insaturados
A maior parte dos ácidos gordos presentes nos fosfolípidos e triacilgliceróis são
insaturados, tendo uma ou mais duplas ligações. Estas ligações estão em configuração cis, não
podendo portanto ser activados pela enol-CoA-hidratase, a enzima que catalisa a adição de
H2O à dupla ligação do Δ2-Enol-CoA durante a -oxidação. Assim, torna-se necessário
recorrer a duas enzimas: uma isomerase e uma redutase.
Qualquer que seja a conformação da cadeia de hidratos de carbono, a -oxidação
ocorre normalmente até à primeira dupla ligação encontrada.
Para melhor compreensão do mecanismo de oxidação iremos ilustrar com dois
exemplos.
O ácido oleico é um ácido gordo mono-insaturado (C18:1) com uma dupla ligação
entre C9 e C10. Após a saída dos primeiros três pares de carbono como acetil-CoA forma-se o
cis-Δ3-dodecenol-CoA. Devido à sua configuração cis torna-se um substracto inapropriado
para a enol-CoA-hidratase, que actua exclusivamente em duplas ligações de configuração
trans.
Recorre-se
então
à
enzima
Δ3-Δ2-enol-CoA-isomerase que vai isomerizar o cis-Δ3-enol-CoA em trans-Δ2-enol-CoA, que
é convertido pela enol-CoA-hidratase em trans-Δ2-dodecenol-CoA. Este pode seguir depois a
via normal da -oxidação, formando mais seis moléculas de acetil-CoA.
Nos casos em que temos um ácido gordo poli-insaturado (como é o caso do ácido
linoleico, de 18 carbonos) outra enzima é necessária. Este ácido gordo tem uma configuração
cis-Δ9- cis-Δ12. O ácido linoleico–CoA segue a sequência inicial da -oxidação, originando
três moléculas de acetil-CoA e um ácido gordo insaturado com a configuração cis-Δ3- cis-Δ6.
Este não pode ser usado pelas enzimas da -oxidação, pois as duplas ligações estão na
posição e configuração erradas. A acção combinada da enol-CoA-isomerase e do 2,4-dienolCoA-redutase vão permitir a reentrada do composto no seguimento da -oxidação, como está
explicado na figura da página seguinte.
A oxidação de ácidos gordos com ligações duplas em carbonos ímpares dá-se pela
cis-Δ2-Enol-CoA-isomerase e em carbonos pares pela cis-Δ2-Enol-CoA-isomerase (que vai
criar uma dupla ligação num carbono ímpar) e pela 2,4-Dienol-CoA-reductase, pelo
mecanismo explicado anteriormente
Catabolismo dos ácidos gordos com número ímpar de átomos de carbono
A -oxidação dos ácidos gordos com número ímpar de átomos de carbono dá-se
exactamente da mesma forma que a -oxidação dos ácidos gordos com número par de átomos
de carbono. No entanto, no último passo desta via metabólica, temos um ácido gordo com
15
cinco carbonos. Ao ser oxidado vai originar uma molécula de acetil-CoA e outra de propionilCoA. O acetil-CoA pode entrar no ciclo do ácido cítrico, mas o propionil-CoA segue outra
via, que envolve três enzimas. Pela propionil-CoA-carboxilase (ligada ao cofactor biotina)
transforma-se em D-metilmalonil-CoA que, através da metilmalonil-CoA-epimerase forma o
seu estereoisómero L. Este sofre depois um rearranjo intramolecular para formar succinilCoA, catalisado pela metilmalonil-CoA-mutase (mais coenzima B12).
Cetogénese
Ao processo que consiste na formação de corpos cetónicos a partir de ácidos gordos
dá-se o nome de cetogénese.
Os corpos cetónicos são produzidos sempre. No entanto, quando se verifica uma
escassez tal de hidratos de carbono que a energia tem de ser obtida através da degradação de
ácidos gordos, a produção de corpos cetónicos aumenta. São produzidos essencialmente nas
mitocôndrias das células hepáticas. Há a quebra da cadeia de carbonos do ácido gordo em
segmentos que contêm apenas 2 carbonos, segmentos esses que se encontram sob a forma de
acetil-CoA. Normalmente o acetil-CoA é oxidado no ciclo dos ácidos tricarboxílicos, mas, no
caso de não haver glicose suficiente, vai-se verificar uma carência de intermediários deste
ciclo pelo que vai ocorrer a acumulação de acetil-CoA.
Assim, dá-se a conversão do acetil-CoA em ácido acetoacético, que posteriormente se
poderá converter em ácido
-
-hidroxibutírico não é um
corpo cetónico, de acordo com as regras da IUPAC (não tem grupo cetona). No entanto,
considera-se como corpo cetónico devido à sua quase instantânea conversão no organismo.
A cetogénese dá-se em 3 passos principais:
Condensação de duas moléculas de acetil-CoA em acetoacetil-CoA, por acção da
tiolase;
Condensação do acetoacetil-CoA com um terceiro acetil-CoA por acção da HMG-hidroxi-
-metilglutaril-CoA (HMG-CoA). O mecanismo
base desta reacção é semelhante ao da condensação do oxaloacetato com acetil-Co-A
para a produção de ácido cítrico, no ciclo de Krebs;
Degradação do HMG-CoA em ácido acetoacético (corpo cetónico) e acetil-CoA por
acção da HMG-CoA liase.
Após a formação do ácido acetoacético, dá-se a sua redução a ácido
-hidroxibutírico
-hidroxibutirato-desidrogenase, sendo que também se pode verificar a
descarboxilação do ácido acetoacético a acetona e CO2.
Os corpos cetónicos são facilmente transportados pelo sangue para as células que os
metabolizam.
16
De forma a poderem ser utilizados pelas células, o ácido acetoacético é activado por
transferência de um acetil-CoA a uma molécula de succinil-
-hidroxibutírico é
normalmente oxidado a ácido acetoacético novamente antes da sua utilização. Nas células, há
a conversão dos corpos cetónicos em acetil-CoA.
A produção de corpos cetónicos é regulada pela disponibilidade de acetil-CoA. Se a
-oxidação dos ácidos gordos no
fígado irá ocorrer a uma taxa elevada, assim como a síntese dos corpos cetónicos a partir do
acetil-CoA resultante. A taxa de produção de corpos cetónicos aumenta se o indivíduo sofre
de subnutrição. No fígado, o acil-CoA formado no citosol pode seguir duas vias distintas.
Pode sofrer
-oxidação por parte das enzimas nas mitocôndrias ou pode ser convertido em
triacilgliceróis ou fosfolípidos, sendo que a via a ser seguida depende da taxa de transferência
da cadeia de acil-CoA para o interior de mitocôndria. Esta é uma importante forma de
regulação dos processos envolvendo o catabolismo de ácidos gordos. A partir do momento em
que entram na mitocôndria, os ácidos gordos serão reduzidos a acetil-CoA. O malonil-CoA é
o primeiro intermediário da biossíntese de ácidos gordos no citosol, sendo que a sua
concentração aumenta sempre que se verifica uma boa “reserva” de hidratos de carbono. A
inibição da carnitina-aciltransferase I pelo malonil-CoA inibe a oxidação dos ácidos gordos
sempre que há um nível de glicose suficiente no fígado. Relativamente às enzimas
-oxidação, sempre que a concentração de NADH é elevada relativamente à
de NAD+
-CoA é inibida; por outro lado, altas concentrações de acetil-CoA
inibem a acção da tiolase.
-oxidação (três ciclos)
acção da isomerase
(passagem de cis-Δ3 para
trans-Δ2
-oxidação (um ciclo)
acção da redutase através do
NADPH
acção da isomerase (a dupla
ligação é transferida para o
segundo carbono)
-oxidação (quatro ciclos)
17
2.7- CATABOLISMO DE PROTEÍNAS E
AMINOÁCIDOS, E UREOGÉNESE
A proteína tem muitas funções importantes no corpo: o sangue necessita das proteínas
para os glóbulos vermelhos, glóbulos brancos e numerosos compostos do plasma; a
imunidade do corpo também depende das proteínas, que são necessárias para a formação dos
anticorpos e dos glóbulos brancos que combatem as doenças; as enzimas e as hormonas (por
exemplo a insulina) também são proteínas, etc. As proteínas podem ser encontradas em
produtos animais como carne, peixe, ovos, leite e seus derivados e em alimentos vegetais
como cereais, grãos e sementes. Todas as fontes de proteínas contêm alguns dos aminoácidos
essenciais, mas em quantidades variadas.
A degradação das proteínas é realizada, inicialmente, por enzimas que são sintetizadas
no estômago, pâncreas e intestino delgado. Tanto o pâncreas como o estômago sintetizam
enzimas sob a forma inactiva (zimogénios) que são activadas por clivagem proteolítica. O
intestino delgado, por outro lado, sintetiza as enzimas já activas.
Uma das enzimas sintetizadas pelo estômago é o pepsinogénio, forma inactiva. A sua
forma activa designa-se por pepsina. Neste processo de activação estão envolvidas as
hormonas: gastrina, histamina e acetilcolina. A gastrina é produzida nas células G ao nível do
antro gástrico que vai ser excretada para o sangue. Quando a concentração de gastrina
aumenta, esta vai actuar ao nível das células parietais do estômago. Estas células fazem com
que haja produção de HCl através de uma bomba H+, k+, ATPase (bomba de protões). Assim,
o pH no interior do estômago diminui o que faz com que o pepsinogénio se transforme em
pepsina (pepsina quebra as ligações peptídicas).
O suco pancreático contém o tripsinogénio e o quimiotripsinogénio, formas inactivas
cujas formas activas designam-se por tripsina e quimiotripsina, respectivamente. A tripsina e
a quimiotripsina hidrolisam polipéptidos, transformando-os em oligopéptidos. Ao nível do
duodeno, o tripsinogénio entra em contacto com a enterocinase, enzima segregada pelas
células da mucosa intestinal, convertendo-se em tripsina, que por sua vez contribui para a
conversão do precursor inactivo quimiotripsinogénio em quimiotripsina, enzima activa.
Existem duas vias para a degradação das proteínas: via proteolítica da ubiquitina
(dependente de ATP) e via lisossomal (independente de ATP). Ambas resultam na quebra das
ligações peptídicas entre os aminoácidos numa proteína – pelas proteases.
Via da ubiquitina
Em geral, na via da ubiquitina, as proteínas são degradadas por um complexo de protease
26S (também designado por proteassoma) que reconhece as proteínas a ser degradas pela
presença de ubiquitina nestas. A ubiquitina é uma proteína, presente em todas as células
eucarióticas, que possui um importante papel na marcação de proteínas para a sua degradação.
Três enzimas (E1, E2 e E3) participam na conjugação de ubiquitina às proteínas. Inicialmente,
a enzima E1 liga-se à ubiquitina tornando-a activa. A ubiquitina activada é então ligada à
enzima E2 e posteriormente à enzima E3 que catalisa a transferência da ubiquitina para a
proteína – alvo. Esta proteína ubiquitinada é depois digerida por um complexo de protease
26S. Esta protease energizada por ATP poupa a ubiquitina, que é reciclada, desdobra a
proteína e digere-a.
18
Via lisossomal
Lisossomas são vesículas repletas de enzimas hidrolíticas em estado inactivo. O perfeito
funcionamento das enzimas lisossómicas depende de um pH próximo de 5. Dentro destas
enzimas destacam-se as catepsinas B, C, D, H, L, às quais se atribui a degradação de proteínas
associadas à membrana celular e de diversas outras proteínas em condições de privação
nutricional (em tecidos como o fígado, o rim e o músculo cardíaco).
Existem dois caminhos alternativos dos quais derivam os materiais a serem digeridos
pelos lisossomas: autofagia e endocitose. A autofagia diz respeito à digestão gradual dos
componentes da própria célula. O primeiro passo da autofagia é um mecanismo de
envolvimento da proteína a ser digerida por uma membrana do retículo endoplasmático, ou
membrana plasmática formando então uma vesícula designada por autofagossomo. Este
autofagossomo funde-se com os lisossomas ocorrendo digestão do seu conteúdo. A
endocitose está envolvida no processo de degradação de materiais estranhos vindos do
extracelular (por meio da endocitose), de proteínas de membrana e componentes celulares
envelhecidos. Esta proteólise é estimulada pelo jejum no fígado. A proteólise ocorre através
de processos selectivos e não selectivos. Entre os não selectivos estão a macroautofagia (fusão
de lisossomas com vacúolos originários do complexo de Golgi e retículo endoplasmático liso)
e a microautofagia (invaginação da superfície lisossomal que leva à produção de vesículas
cujo conteúdo proteico sofre degradação no interior do lisossoma).
Ao processo contínuo de síntese e degradação das proteínas dá-se o nome de reciclagem
ou renovação proteica. Em geral um adulto degrada 1-2% das suas proteínas por dia. Cerca
de 75-80% dos aminoácidos libertados são usados para nova síntese proteica e os restantes
20-25% são degradados (e não armazenados).
A renovação proteica permite a síntese de proteínas adequadas assim como a degradação
de proteínas desnecessárias. Para além disso, protege as células de acumulação de proteínas
anormais (como por exemplo erros na síntese proteica ou desnaturação espontânea).
Proteínas
Síntese
Degradação
Aminoácidos
É de notar que a degradação das proteínas em resposta ao stress, deficiências nutritivas
e cansaço pode ser suprimida ou promovida. Em resposta ao stress, o corpo segrega a
epinefrina, a noreepinefrina, o cortisol e outras hormonas. Os glicocorticóides (tais como o
cortisol) têm uma acção catabólica, suprimindo, assim, a síntese de proteínas e promovendo a
degradação destas em aminoácidos. Este processo é necessário para neutralizar o stress. No
entanto, se o processo for prolongado, o catabolismo resultante é muito prejudicial ao corpo
que pode resultar na supressão do sistema imunitário, dos orgãos digestivos, das hormonas de
crescimento e de outros sistemas importantes do corpo.
Quando um corpo apresenta deficiências nutritivas, ou seja, quando não pode metabolizar
correctamente açúcares, hidratos de carbono e gorduras que participam nos ciclos da glicólise
e ciclo de krebs haverá digestão das proteínas endógenas a fim de produzir energia. O uso da
proteína para a obtenção de energia não é necessariamente económico para o corpo, porque a
manutenção, o crescimento, e o reparo dos tecidos são comprometidos para se encontrar
necessidades de energia. Também, a fatiga e outras condições da saúde podem causar um
estado catabólico superior.
Os aminoácidos, constituintes das proteínas, podem ser usados como precursores de
moléculas biológicas azotadas, pois têm na sua constituição um grupo amina. O excesso de
aminoácidos da dieta não é armazenado nem excretado mas sim convertido em piruvato,
19
oxalacetato e α-cetoglutarato. Consequentemente, os aminoácidos podem também ser
considerados precursores da glicose, ácidos gordos e corpos cetónicos. Deste modo, podem
ser considerados “compostos energéticos”.
O maior local de degradação dos aminoácidos é o fígado e neste processo está envolvido
a eliminação do grupo amina dos aminoácidos a ser degradados. As principais reacções
envolvidas na eliminação do grupo amina são a transaminação e a desaminação oxidativa. A
uma transaminação acoplada a uma desaminação oxidativa dá-se o nome de
transdesaminação. Na transaminação há transferência do grupo amina de um aminoácido
para um α-cetoácido, originando o α-cetoácido e aminoácido correspondentes:
aminoácido A + cetoácido B → aminoácido B + cetoácido A
Na desaminação oxidativa há remoção do grupo amina de um aminoácido, catalisada
por uma aminotransferase com redução de NAD+ (ou NADP+) a NADH + H+(ou NADPH +
H+):
aminoácido + NAD(P)+ + H2O → cetoácido + NAD(P)H + H+ + NH4+
As aminotransferases que catalisam este tipo de reacções são específicas para cada tipo
de aminoácidos originando α-cetoácidos correspondentes.
Assim, no decurso do seu catabolismo os aminoácidos perdem os seus átomos de
azoto que, na sua maioria, são incorporados na ureia e excretados na urina.
O amoníaco (NH3) forma-se em todos os tecidos, mas como é um composto
extremamente tóxico para o organismo é incorporado em alguns compostos menos tóxicos. O
amoníaco pode ser transportado para o fígado sob a forma de glutamato, glutamina ou
alanina. Em meio aquoso o amoníaco (NH3) encontra-se sob a forma de ião amónio (NH4+).
O amoníaco é obtido a partir dos aminoácidos quando estes são necessários como
percursores da glicose ou para fornecer energia. O metabolismo bacteriano no lúmen
intestinal também é uma fonte importante de amoníaco, sendo absorvido e transportado para o
fígado.
A glutamato desidrogenase catalisa a reacção em que se forma glutamato a partir da
incorporação de amoníaco no -cetoglutarato. A reacção contrária é catalizada pela mesma
enzima. O glutamato é um dos aminoácidos que entram nas transaminações e desaminações
oxidativas. A glutamato desidrogenase é regulada alostericamente por nucleótidos de purinas.
Quando os níveis de ADP e GDP são elevados, é nessessário a oxidação de aminoácidos para
a obtenção de energia e a glutamato desigrogenase aumenta a sua actividade no sentido de
degradar glutamato. Por outro lado quando os níveis de ATP e GTP são elevados estes são
activadores alostéricos da síntese de glutamato.
A glutamina representa metade dos aminoácidos em circulação e o seu grupo amina é
um doador de azoto para várias classes de moléculas como as bases púricas e o grupo amina
da citosina. Na presença da glutamina sintase e ATP o amoníaco é incorporado no glutamato
formando glutamina. Esta reacção ocorre em duas etapas. Numa primeira fase, o ATP doa um
grupo fosforil ao glutamato formando-se um composto intermediário (γ-glutamil-fosfato). Na
segunda fase, o amoníaco reage com este composto intermediário deslocando o fosfato
inorgânico para produzir glutamina.
O amoníaco produzido na degradação dos aminoácidos nos músculos também é
transportado para o fígado sob a forma de alanina usando o ciclo glicose-alanina. Nos
músculos o amoníaco reage com o -cetoglutarato na presença da glutamato- desidrogenase
formando glutamato, que por sua vez na presença de alanina-transaminase transfere o seu
grupo amina para o piruvato formando alanina. A alanina no fígado transfere o seu grupo
amina para o -cetoglutarato por intermédio da alanina-transaminase.
Em condições de necessidade energética as proteínas que estão nas células musculares
são degradadas e os grupos amina são transferidos para produzir glutamina e alanina e
transportados para o rim e fígado.
20
O fígado é o principal destino da glutamina e da alanina do sangue, onde o amoníaco é
libertado pela alanina aminotransferase, glutaminase e glutamato desidrogenase. A última
também produz NADH e -cetoglutarato, um intermediário do glicogénio.
De uma forma geral, dos processos de degradação de aminoácidos resultam grupos
amina, que não sendo utilizados pelo organismo para sintetizar novos aminoácidos ou outros
produtos azotados, são modificados dando origem a um produto final que será excretado. O
grupo amina removido a partir do aminoácido degradado forma iões amónio. O ião amónio é
extremamente tóxico para os organismos da grande maioria dos mamíferos terrestres e por
isso tem que ser convertida num composto não – tóxico, e portanto tolerável, por estes. Este
composto designa-se ureia. A ureia é produzida a partir de várias reacções sequenciais que
constituem o Ciclo da Ureia (Ureogénese ou Ciclo de Krebs-Henseleit) e posteriormente
excretada na urina.
A formação de ureia - NH2CONH2 - ocorre nas células hepáticas, principais células
do fígado, a partir de dois compostos inorgânicos, dióxido de carbono – CO2 e iões amónio –
NH4+. Estes iões têm origem na remoção de grupos amina. Para a remoção dos grupos amina
existem diferentes processos possíveis. Num processo está envolvida uma transdesaminação:
a transaminação envolvida na transferência do grupo amina de um aminoácido para o ceglutarato originando glutamato e por outro lado, a desaminação oxidativa catalisada pela
glutamato - desidrogenase, havendo remoção do grupo amina do glutamato. O grupo amina
resultante entra no ciclo da ureia. Um segundo processo inclui duas reacções de
transaminação. A primeira destas é a transferência do grupo amina para o -cetaglutarato
resultando a formação do glutamato. A segunda, catalisada pela aspartato aminotransferase, é
a transferência do grupo amina do glutamato para o oxaloacetato resultando então a formação
de aspartato. O Aspartato formado entra no ciclo da ureia pela condensação com a citrulina.
Desta forma, um segundo grupo amina entra no ciclo, dando portanto origem a um segundo
átomo de azoto que será utilizado na formação de ureia. Uma pequena parte dos iões amónio
utilizada no ciclo da ureia pode ser originada pela oxidação de aminoácidos pelas bactérias
existentes na flora intestinal e transportada até ao fígado pela veia porta.
O ciclo da ureia inclui cinco reacções principais que permitem a síntese do composto
orgânico ureia a partir de dois compostos inorgânicos, dióxido de carbono – CO2 e iões
amónio – NH4+. As duas primeiras reacções do ciclo envolvido ocorrem nas mitocôndrias e as
restantes três no citosol das referidas células.
O ciclo inicia-se com a formação do Carbomoil Fosfato. Esta reacção é catalisada pela
carbomoil fosfato sintase I (CPSI) e é irreversível, constituindo, assim, um local importante
de regulação do ciclo. Esta reacção implica o consumo de duas moléculas de ATP. Segue-se a
a formação de citrulina na qual se dá a transferência do grupo carbomoil para a ornitina pela
ornitina transcarboximoilase. A ornitina é uma molécula transportadora que permite a
progressão do ciclo pois auxilia no transporte de citrulina do interior da mitocôndria para o
citosol. Segue-se a síntese de argininosuccinato, catalisada pela argininosuccinato sintetase, e
onde ocorre condensação da citrulina com o aspartato. Esta reacção é desencadeada pela
clivagem de ATP, da qual resultam AMP e pirofosfato. O pirofosfato é rapidamente
hidrolisado originando dois fosfatos inorgânicos. Existe, assim, consumo de dois equivalentes
de ATP. A reacção que se segue é a clivagem de argininosuccinato, catalizada pela
ariginosuccinato liase, da qual resultam fumarato e arginina. Por fim tem-se a clivagem de
arginina da qual resultam a ornitina e a ureia. A enzima que catalisa esta clivagem designa-se
arginase. Esta enzima existe exclusivamente no fígado o que torna a produção de ureia
exclusiva a este órgão. Existem, ainda, transportadores específicos para a ornitina na
membrana mitocondrial interna pelo que depois de originada esta é transportada para o
interior da mitocôndria permitindo iniciar-se um novo ciclo. A ureia formada é transportada
pelo sangue até aos rins para posteriormente ser excretada na urina.
O fumarato formado no ciclo da ureia é convertido em malato estando nesta reacção
envolvida a enzima fumarase. No citosol, o malato pode por um lado ser convertido em
oxaloacetato que por sua vez pode ser convertido em aspartato ou por outro lado ser
transportado para o interior da mitocôndria e aí entrar no ciclo do ácido cítrico. Os NADH
21
formados em ambas as situações podem ser oxidados pela cadeia transportadora de electrões
dando, cada molécula de NADH, origem a 2,5 moléculas de ATP.
Por outro lado, o fumarato é também um interveniente no ciclo do ácido cítrico. Os ciclos
estão assim interligados. Apesar disto, estes ciclos ocorrem independentemente e a
comunicação entre ambos depende do transporte de intermediários entre a mitocôndria e o
citosol. Várias enzimas do ciclo do ácido cítrico incluindo a fumarase (fumarato hidratase) e a
malato desidrogenase estão presentes como isoenzimas no citosol. O fumarato originado a
partir da síntese no citosol de arginina pode ser convertido em malato no citosol e estes
intermediários podem ser depois metabolizados no citosol ou transportados para o interior da
mitocôndria para serem usados no ciclo do ácido cítrico. O aspartato formado na mitocôndria
por transaminação entre o oxaloacetato e o glutamato pode ser transportado para o citosol
onde funciona como dador de azoto no ciclo da ureia.
A regulação do ciclo da ureia pode ser considerada a dois níveis. A regulação principal
do ciclo da ureia é realizada pelo N-acetil glutamato (formado a partir da acetil-CoA e do
glutamato) que activa alostericamente a carbomoil fosfato sintase I (CPSI). Tal como já foi
mencionado esta enzima cataliza a primeira reacção do ciclo sendo por isso determinante na
sua regulação global. Se for seguida uma dieta rica em proteínas, os aminoácidos excedentes
são desaminados. Disto resulta um aumento da concentração de glutamato e portanto de Nacetil glutamato. O N-acetil glutamato activa a CPSI, e desta forma, promove o desenrolar do
ciclo da ureia com o objectivo de compensar o excesso de azoto existente. Existe um outro
tipo de regulação do ciclo, só que este a longo prazo. Sabe-se que alterações na dieta podem
induzir ou inibir a transcrição das enzimas envolvidas no ciclo da ureia. Por exemplo, em
jejum, há um aumento da degradação das proteínas constituintes de alguns tecidos o que
induz a síntese de enzimas intervenientes no ciclo da ureia, de forma a compensar o excesso
de ião amónio originado.
22
2.9- Gliconeogénese
Na ausência de glicose, a manutenção da glicemia faz-se a partir da síntese de glicose
a partir de percursores não glicídicos. Define-se gliconeogénese como a formação de glicose
a partir de material não glicídico.
O fígado e os rins são denominados sítios de gliconeogénese, e compensam a sua fraca
capacidade glicolítica com a elevada capacidade gliconeogénica (processos realizados em
células diferentes). Nestes sítios os precursores seguem vias especiais, dando genericamente e
directa ou indirectamente piruvato, que segue a via comum.
São os aminoácidos, os lípidos e o lactato que seguem as vias especiais. Nos
aminoácidos a via faz-se apenas utilizando aminoácidos glicoformadores, que a partir de
aminações ou transaminações originam piruvato, α-cetoglutarato, oxaloacetato ou SuccinilCoA. Nos lípidos, apenas o glicerol se converte em dihidroxicetona fosfato, um interveniente
da glicólise/gliconeogénese.
O lactato, como se sabe, é produzido constantemente nos eritrócitos e também nos
músculos sob exercício intenso. Como não existem nestes locais as enzimas necessárias para
fazer o processo inverso, então tem que ser transportado até ao fígado (ou rim) que através do
ciclo de Cori é oxidado a piruvato.
A via comum ou final é por assim dizer o processo inverso da glicólise, é em tudo
igual, excepto em três reacções, as de regulação.
De uma maneira ou de outra, todos os produtos das vias especiais dão origem a
piruvato, que é então convertido em oxaloacetato que origina fosfoenolpiruvato. O que sucede
é que o oxaloacetato é formado dentro da mitocôndria e o fosfoenolpiruvato fora desta, então,
como o oxaloacetato não consegue atravessar a membrana, tem
de ser convertido em malato para poder ser transportado para
fora da mitocôndria e voltar a oxaloacetato para originar
fosfoenolpiruvato,
no
primeiro
e
segundo
passos
da
gliconeogénese (um dos passos de regulação).
Pode-se ver no esquema a representação das duas vias
antagónicas
glicolise/gliconeogénese,
e
as
enzimas
intervenientes.
A regulação deste processo metabólico é feita pelas
enzimas dos passos de regulação. As duas primeiras, a piruvato
carboxilase
e
a
fosfoenolpiruvato
carboxicinase
são
estimuladas pelo acetil-CoA, e são passos com grande energia
de activação, sendo altamente desfavoráveis (como se pode ver
23
gasta-se um ATP e um GTP, moléculas que cedem a energia necessária para ultrapassar esse
obstáculo). Não é por acaso que os passos de maior energia de activação são os de regulação,
faz sentido que assim seja, de modo a garantir a permanência de apenas um sentido de cada
vez.
A frutose-1,6-bisfosfatase é estimulada pelo citrato e inibida pelo AMP (que estimula
a glicólise, fazendo sentido inibir a gliconeogénese) e pela frutose-2,6-bifosfato, molécula
controlada pelo sistema hormonal insulina/glicagina. Por ultimo, e na regulação da ultima
reacção da gliconeogénese, encontramos a glicose-6-fosfatase que apesar de não ser
controlada alostericamente varia aproximadamente de uma maneira linear com a concentração
de substrato.
Como se pode ver pelo esquema, a gliconeogénese é um processo que requer muita
energia, de modo que é um processo de recurso quando não há mais nada que forneça glicose
de uma maneira mais fácil, no sentido de aumentar a glicemia.
Catabolismo dos aminoácidos glicogénicos
Quanto ao destino dos produtos que advêm do seu metabolismo, os aminoácidos
podem dividir-se em glicogénicos, que originam metabólitos que são incorporados como
intermediários no Ciclo de Krebs e no metabolismo da glicose (piruvato, alfa-cetoglutarato,
succinil-CoA, fumarato e oxaloacetato), e cetogénicos, que originam metabólitos que são
incorporados no metabolismo dos lípidos, podendo formar ácidos gordos ou corpos cetónicos
(acetil-CoA ou acetoacetil-CoA).
É importante referir que existem aminoácidos que, no decurso do seu catabolismo,
originam acetil-CoA e intermediários do Ciclo de Krebs ou da glicólise, sendo classificados
como simultaneamente glicogénicos e cetogénicos. Actualmente, a leucina é tido, por muitos,
como o único aminoácido exclusivamente cetogénico.
De seguida, ir-se-á explicar mais a fundo o catabolismo de alguns dos aminoácidos
glicogénicos. Não é demais referir que o facto de os aminoácidos poderem gerar piruvato e/ou
intermediários do ciclo de Krebs e/ou acetil-CoA permite compreender que, ao serem
oxidados a CO2, podem contribuir para a síntese de ATP sendo, a par com os glícidos e os
lípidos, compostos potencialmente energéticos.
Para se compreenderem os mecanismos presentes no catabolismo dos aminoácidos, é
necessário ter, pelo menos, estas duas noções básicas:
Transaminação - consiste na transferência reversível do grupo amina de um
aminoácido para um alfa-cetoácido, na presença da transaminase, produzindo o
aminoácido correspondente ao alfa-cetoácido, e o alfa-cetoácido correspondente ao
24
aminoácido original. Geralmente, o aceitador do grupo amina é o alfa-cetoglutarato,
que é convertido em glutamato
Desaminação oxidativa - por acção da glutamato desidrogenase, dá-se a remoção do
grupo amina, sob a forma de ião amónio livre, a partir do glutamato proveniente,
sobretudo, das reacções de transaminação. O NAD+ funciona como aceitador de
electrões, isto quando o pH é de 9.0, pois caso este suba para 9.5, o aceitador de
electrões será o NADP+.
- A asparagina é hidrolisada, por acção da asparaginase, originando aspartato e iões amónio.
Seguidamente,
o
aspartato
sofre
uma
transaminação
(aspartato
+
alfa-cetoácido
glutamato + oxaloacetato) originando oxaloacetato.
- A serina sofre uma desaminação, originando piruvato, sendo que o grupo amina é libertado
como ião amónio, ao invés de ser transferido para outro composto.
- A cisteína é convertida a piruvato por um processo que liberta ião amónio e enxofre, que
advém da oxidação do grupo tiol da cisteína. Existe outro processo alternativo de catabolismo
da cisteína, processo este em que não há perda do grupo azotado formando-se, em vez de
piruvato, taurina (que é, em última análise, eliminada na urina).
25
- Aquando da transaminação do aspartato e da alanina forma-se, para além de,
respectivamente, oxaloacetato e piruvato, glutamato.
- O glutamato pode, por desaminação, originar alfa-cetoglutarato (intermediário do Ciclo de
Krebs) e, por outro lado, pode também dar origem a alfa-cetoglutarato por acção da glutamato
desidrogenase. Esta reacção dá também origem ao ião amónio.
O alfa-cetoglutarato pode seguir dois caminhos distintos, sendo utilizado como intermediário
no Ciclo de Krebs ou, pelo contrário, numa outra transaminação.
- A treonina é um exemplo de aminoácido simultaneamente glicogénico e cetogénico, uma
vez que forma acetil-CoA e glicina. A acetil-CoA é precursora de corpos cetónicos e a glicina
é potencialmente glicogénica, dado que pode ser convertida a serina por acção da serinahidroximetiltransferase.
(ver, na apresentação, a figura-resumo da relação entre o catabolismo dos aminoácidos e o
Ciclo de Krebs / Gliconeogénese.)
Os iões amónio, formados por transaminação/desaminação oxidativa e por outras reacções
são exportados dos tecidos extra-hepáticos para o fígado através de dois mecanismos de
26
transporte: a síntese de glutamina e o ciclo glicose-alanina (sendo que e dará mais importância
ao primeiro, uma vez que é o mais “utilizado”).
- Síntese da Glutamina:
Primeira reacção – síntese da glutamina, por parte da maioria dos tecidos, a partir do
glutamato, como forma de armazenamento temporário não tóxico e transporte de amónio para
o fígado ou para os rins.
Segunda reacção – a glutamina é hidrolisada, no fígado e rim, a glutamato e amónia, pela
acção da enzima glutaminase.
-
A partir da ultima reacção:
No fígado – o NH3 é utilizado na síntese da ureia;
No rim – o glutamato sofre desaminação oxidativa, dando origem
a alfa-cetoglutarato, sendo excretadas, na urina, dois iões amónio para cada glutamina
transformada em alfa-cetoglutarato. Nos túbulos renais, o amoníaco é protonado a iões
amónio, que neutralizam os ácidos metabólicos na urina.
Metabolismo do glicogénio
A glicose pode ser armazenada, no nosso organismo, sob a forma de glicogénio.
Este é, então, um polissacarídeo originado por polimerização da glicose. O glicogénio é muito
ramificado, possuindo ligações glicosídicas α-1,4 ao longo de um mesmo ramo, e ligações α1,6 nos pontos de derivação de novos ramos, assim como terminais não redutores, na sua
maioria.
As reservas de glicogénio estão centradas no fígado e no tecido muscular esquelético,
onde este aparece sob a forma de grânulos citosólicos, juntamente com a generalidade das
enzimas necessárias ao seu metabolismo.
No que diz respeito a esse mesmo metabolismo, têm-se os processos de glicogenólise,
catabólico, que visa a obtenção de glicose-6-fosfato (G-6-P) a partir do glicogénio, e de
glicogénese, síntese / alongamento de moléculas de glicogénio, anabólico.
Na glicogenólise, por acção da glicogénio-fosforilase, um resíduo de glicose é
removido de um terminal não redutor da cadeia de glicogénio. A ligação α-1,4 é atacada por
27
um fosfato inorgânico, originando-se glicose-1-fosfato (G-1-P), e encurtando-se a cadeia –
Fosforólise. A actividade desta enzima é sucessivamente repetida, e cessada quando esta
atinge um ponto à distância de 4 resíduos de uma ramificação (ligação α-1,6). Aí, é necessária
a enzima desramificante, para se poder continuar o processo.
Em primeiro lugar, a enzima desramificante actua como transferase, transferindo um
bloco de 3 resíduos de glicose para um terminal não redutor, formando-se uma ligação α-1,4.
Posto isto, o único resíduo restante na ramificação é removido pela acção de glicosidase desta
enzima, soltando-se uma molécula de glicose simples, e assim se obtém de novo uma cadeia
apelativa à actividade da fosforilase.
Segue-se a acção da fosfoglicomutase, que catalisa a reacção reversível entre a G-1-P
e a G-6-P. Inicialmente fosforilada, a enzima cede o seu grupo fosfato à G-1-P, formando-se
glicose-1,6-difosfato. Esta cede o seu fosfato-1 de novo à enzima, que se fosforila novamente,
originando-se G-6-P.
No tecido muscular esquelético, o glicogénio é de consumo local – A G-6-P obtida
entra directamente na glicólise, que conduzirá à produção de energia necessária à contracção
muscular. Já no fígado, o objectivo último é, sim, a libertação de glicose para o sangue,
nomeadamente em períodos em que a glicémia tende a baixar, como em jejum, para
restabelecer os níveis desta. Existe então, apenas no fígado, a G-6-fosfatase. Esta enzima é
proteína integrante da membrana do retículo endoplasmático, tendo o seu centro activo na
face interna da mesma membrana. Tal facto implica a existência de transportadores
específicos para mover a G-6-P para o interior do retículo, e para conduzir os produtos da sua
hidrólise, glicose e fosfato inorgânico, de volta ao citosol. A glicose é então encaminhada à
corrente sanguínea por outro transportador (GLUT2).
Quando se verifica uma elevada glicémia, ou em períodos de repouso, no músculo, todo este
processo é cessado e tem início a glicogénese.
A glicogénese inicia-se com a acção inversa da fosfoglicomutase, obtendo-se, a partir
de G-6-P, G-1-P, que por sua ver reage com o UTP para originar UDP-glicose, o substrato do
alongamento da molécula de glicogénio. A enzima glicogénio-sintase vai catalisar este
processo, transferindo resíduos de glicose da UDP-glicose para um terminal não redutor do
glicogénio, formando-se uma ligação α-1,4. A enzima ramificante vai, depois da adição de
diversos resíduos pela sintase, transferir um fragmento de 6-8 resíduos
para longe do
terminal, formando-se uma ligação α-1,6, e uma nova ramificação.
Em resposta à incapacidade da glicogénio-sintase de sintetizar uma molécula de
glicogénio a partir do zero, surge a glicogenina, que para além de ser a molécula onde os
primeiros resíduos de glicose se vão ligar, é também a enzima que catalisa estas mesmas
28
ligações, com a sua actividade intrínseca de glicosiltransferase. Chegando aos 8 resíduos de
comprimento, inicia-se então a acção da glicogénio-sintase.
Sumarizando a regulação não hormonal dos processos metabólicos do glicogénio,
temos, a regulação por fosforilação reversível das enzimas glicogénio-fosforilase e
glicogénio-sintase, da responsabilidade das cinases (fosforilação) e da fosfatase-1
(desfosforilação). A fosforilação vai activar a fosforilase, e inactivar a sintase, favorecendo a
glicogenólise, enquanto que a desfosforilação tem o efeito oposto, contribuindo para a
ocorrência de glicogénese. As cinases e a fosfatase-1 podem ainda ser, também, reguladas por
fosforilação.
No músculo, a glicogénio-fosforilase é ainda activada e inibida alostericamente pelo
AMP e ATP, respectivamente, e a cinase da fosforilase é activada alostericamente pelo
complexo cálcio-calmodulina. A glicogénio-sintase é activada por ligação à G-6-P, uma vez
que esta facilita a sua desfosforilação.
Os transportadores de glicose do fígado, GLUT2, permitem um equilíbrio entre a
concentração de glicose no sangue e nos hepatócitos. A glicose, quando em grande
quantidade, vai activar a fosfatase-1, que por sua vez inactiva a fosforilase, inibindo-se a
degradação do glicogénio.
29
2.10- Síntese de Ácidos Gordos
a) Biossíntese de ácidos gordos
Os ácidos gordos são sintetizados sempre que há excesso calórico na dieta, em
diversos órgãos, como sejam o fígado, cérebro, rim, pulmão, tecido adiposo, entre outros.
O local desta síntese é no citoplasma da célula, sendo que a principal origem do
carbono para esta via é proveniente dos glícidos da dieta alimentar. A glicose é convertida em
piruvato (via glicólise), que entra para a mitocôndria, formando acetil-CoA e oxaloacetato.
Como a síntese de ácidos gordos ocorre no citoplasma, ao passo que a síntese de acetil-CoA
ocorre na mitocôndria é necessário transportar a acetil-CoA para o citoplasma. Isto é feito
pelo sistema de transporte dos ácidos tricarboxílicos, também chamado ciclo do citrato: o
citrato formado na mitocôndria por condensação do acetil-CoA com oxaloacetato difunde-se
para o citoplasma, onde é clivado pela citrato-liase em acetil-CoA ( fonte dos carbonos
utilizados na síntese) e oxaloacetato, que é depois reduzido a malato pela malato
desidrogenase, que se pode difundir de volta para a mitocôndria ou originar piruvato (redução
do malato a piruvato pela enzima málica, que pode ser uma fonte de NADPH, como é
referido mais à frente), que também se difunde para a mitocôndria.
1º Passo – carboxilação da acetil-CoA a malonil-CoA
É um processo irreversível, não ocorrendo, portanto, na β-oxidação. A reacção é
catalizada pela acetil-CoA carboxilase. Esta proteína, que na célula animal constitui um
polipéptido multifuncional, necessita da biotina e é constituída por 3 regiões funcionais:
– proteína transportadora de biotina, que está ligada covalentemente à biotina por uma
ligação amida;
– biotina carboxilase, responsável pela activação do CO2 (proveniente do ião
bicarbonato), e sua transferência para a biotina, numa reacção dependente de ATP;
– transcarboxilase, responsável pela transferência do CO2 da biotina para a acetilCoA, produzindo o malonil-CoA.
Complexo multienzimático da síntese de ácidos gordos
Este complexo é constituído por um dímero, em que cada monómero consiste numa
cadeia polipeptídica que contém as setes actividades enzimáticas da síntese: β-cetoacil-ACP
sintetase, acetil-CoA-ACP transacetilase, malonil-CoA-ACP transferase, tioesterase, βcetoacil-ACP redutase, enoil-ACP redutase e β-hidroxiacil-ACP desidratase.
30
Síntese de ácidos gordos
A ACP (proteína transportadora de acil) é uma pequena proteína que contém um grupo
prostético, 4’-fosfopantetaína. O grupo –SH pertencente a este grupo prostético é o local de
ligação do grupo malonilo (CoA é libertada) durante a síntese, através da malonil-CoA-ACP
transferase. O grupo acetilo (proveniente da acetil-CoA, CoA é libertada) é necessário para a
primeira reacção do primeiro ciclo da síntese, ligando-se ao grupo – SH da β-cetoacil-ACP
sintetase, ligação esta promovida pela acetil-CoA-ACP transacetilase.
As cadeias de ácidos gordos são formadas a partir de repetidas sequências (ciclos) de 4
passos:
– 1ª reacção: condensação do grupo acetilo e do grupo malonilo formando o
acetoacetil-ACP, através da β-cetoacil-ACP sintetase; por cada passagem pelo ciclo,
a cadeia é aumentada em 2 carbonos e é libertada uma molécula de CO2 do grupo
malonilo, a qual foi adicionada aquando da carboxilação da acetil-CoA a malonilCoA;
– 2ª reacção: redução do acetoacetil-ACP, formando-se o β-hidroxibutiril-ACP,
sendo catalisada pela cetoacil-ACP reductase; o NADPH é oxidado a NADP+;
– 3ª reacção: remoção do elemento água (desidratação do β-hidroxibutiril-ACP),
formando-se o trans-Δ2-butenoil-ACP, através da β-hidroxiacil-ACP desidratase;
forma-se uma dupla ligação entre o C2 e o C3;
– 4ª reacção: a dupla ligação do trans-Δ2-butenoil-ACP é reduzida (saturada),
formando-se butiril-ACP, reacção catalisada pela enoil-ACP reductase. Aqui o
dador de electrões, tal como na 2ª reacção, é o NADPH, que é oxidado a NADP+.
Posteriormente, o grupo butiril-ACP é transferido do grupo –SH do ACP para o grupo
–SH da β-cetacil-ACP sintetase, de forma a se poder ligar mais um grupo malonilo à ACP, e
assim se poder reiniciar um novo ciclo de 4 reacções.
As reacções de condensação e redução param, geralmente, após 7 ciclos, com a
formação do composto saturado palmitil-ACP (16 carbonos). Numa reacção de hidrólise
(quebra da ligação entre o palmitato e a ACP), catalisada pela tioesterase, ocorre a libertação
do palmitato do ACP.
Reacção geral do processo:
8 acetil-CoA + 7ATP + 14NADPH + 14H+
Palmitato + 8CoA + 7ADP + 7Pi +
14NADP+ + 6H2O
31
A activação prévia dos ácidos gordos consiste numa reacção catalisada pela acil-CoA
sintetase: os ácidos gordos reagem com o ATP e CoA gerando-se como produtos AMP,
pirofosfato (PPi) e acil-CoA (ácido gordo + CoA + ATP → acil-CoA + AMP + PPi).
Fontes de NADPH
As principais fontes de NADPH para as reduções ocorridas durante a síntese são:
– a enzima málica, na reacção de conversão do malato a piruvato;
– a via das fosfopentoses, que decorre igualmente no citoplasma;
– a enzima desidrogenase isocítrica, que catalisa a formação α-cetoglutarato a partir
do isocitrato, durante o ciclo de Krebs.
b) Alongamento e redução dos ácidos gordos sintetizados
O destino metabólico do palmitato formado é ser tioesterificado com a CoA formando
palmitil-CoA (acil-CoA sintetase) que pode estar na origem de triacilgliceróis e outros lípidos
(esterificação, isto é, os ácidos gordos reagem com álcoois produzindo ésteres; é
particularmente importante no tecido adiposo, fígado, glândula mamária activa e intestino
delgado), de ácidos gordos com maior número de carbonos (alongamento), de ácidos gordos
insaturados
(dessaturação)
ou
sofrer,
-oxidação (os ácidos gordos
transportados para a mitocôndria pela carnitina
são, através da
-oxidaçao, degradados em
acil-CoA e acetil-CoA, que por sua vez são
intervenientes no ciclo de Krebs). Neste
trabalho apenas vamos focar o alongamento e a
dessaturação.
Alongamento
O palmitato, que é o principal produto
da síntese de ácidos gordos nas células animais,
é o percursor das cadeias longas de ácidos
gordos.
Pode
ser
alongado
para
formar
estearato (18:0) (estearato (18C) forma-se por
adição de 2 átomos de carbono ao palmitato
(16C) ) ou mesmo maiores ácidos gordos
saturados, por sucessivas adições de unidades
32
de 2 carbonos (do malonil-CoA) à acil-CoA, através da acção dos sistemas de alongamento
dos ácidos gordos presentes no retículo endoplasmático liso e na mitocôndria.
O sistema de alongamento mais activo do reticulo endoplasmático estende a cadeia de
palmitoil-CoA, formada por 16 carbonos, a mais 2 carbonos, formando o estearoil-CoA.
O palmitato é o percursor do estearato e dos ácidos gordos saturados de cadeia longa,
como o palmitoleato e o oleato. Os mamíferos não podem converter oleato em linoleato ou
em α-linolenato, os quais são fundamentais na dieta alimentar, pois são ácidos gordos
essenciais. A conversão do linoleato a outros ácidos gordos polinsaturados e eicosanóides está
esboçada na figura. Os ácidos gordos insaturados estão indicados com o número de carbonos
seguidos do número de ligações duplas e este seguido da posição dessas ligações duplas.
Dessaturação
Cada dessaturação consiste em acrescentar 1 ligação dupla ao ácido gordo, mantendo
o número de carbonos. Ou seja o ácido gordo deixa de ser saturado e passa a ser insaturado.
Reparemos na dessaturação do palmitato a palmitoleato: mantém o mesmo número de
carbonos (16) mas acrescenta uma ligação dupla na posição 9.
O palmitato e o estearato são os percursores dos ácidos gordos monoinsaturados mais
comuns do tecido animal: o palmitoleato, 16:1(Δ9), e o oleato, 18:1(Δ9). A ligação dupla é
introduzida dentro da cadeia do ácido gordo por uma reacção de oxidação catalizada pela acilCoA dessaturase, que é uma oxidase. Nesta reacção, dois substratos diferentes, o ácido gordo
e o NADH ou NADPH, são simultaneamente oxidados. O trajecto do fluxo do electrão inclui
um citocromo b5 e uma flavoproteína (citocromo b5 redutase), os quais, tal como a acil-CoA
desaturase, estão no reticulo endoplasmático.
Nos mamíferos não há síntese de ácidos gordos polinsaturados, apenas de
monoinsaturados: o palmitoleato e o oleato. Os hepatócitos dos mamíferos podem introduzir
ligações duplas na posição Δ9 dos ácidos gordos, mas não podem introduzir ligações duplas
adicionais entre o carbono 10 e o metil terminal. Portanto, os mamíferos não conseguem
sintetizar linoleato, 18:2(Δ9,12), ou α-linolenato, 18:3(Δ9,12,15).
As plantas, contudo, podem sintetizar tanto ácidos gordos polinsaturados como ácidos
gordos monoinsaturados. Pelo facto de serem os percursores necessários para a síntese de
outros produtos, o linoleato e o linolenato são ácidos gordos essenciais para os mamíferos,
mas como estes não os conseguem sintetizar, devem ser obtidos a partir de vegetais. Uma vez
ingerido, o linoleato deve ser convertido em certos ácidos polinsaturados, particularmente, o
γ-linolenato, o eicosatirenoato e o araquidonato. O araquidonato, 20:4(Δ5,8,11,14) é um
percursor essencial à regulação dos lípidos, dos eicosanóides.
33
Regulação da Síntese de ácidos gordos
O citrato forma-se dentro da mitocôndria, no ciclo de Krebs, quando o oxaloacetato reage
com o acetil-CoA e por acção da enzima citrato sintase forma o citrato. Quando a
concentração de acetil-CoA e ATP, na mitocôndria, aumenta, o citrato é transportado para
fora da mitocôndria. Aí, vai activar a enzima acetil-CoA carboxilase, que por sua vez vai
catalisar a formação do malonil-CoA. Portanto, a activação desta enzima constitui a etapa
limitante da biossíntese de ácidos gordos.
O citrato inibe a actividade da fosfofrutocinase-1, reduzindo o fluxo de carbono
através da glicólise. O citrato é um activador alostérico, uma vez que quando se liga à enzima
acetil-CoA carboxilase (num sítio diferente do seu centro activo), vai provocar uma alteração
conformacional desta enzima, o que faz aumentar a actividade enzimática. Portanto, o Vmax
da reacção aumenta. O citrato tem um papel muito importante no metabolismo celular na
medida que impede que o combustível metabólico seja consumido e em vez disso é
armazenado como ácidos gordos.
Para além do citrato, a acetil-CoA carboxilase pode ser activada pela hormona insulina
que vai provocar uma desfosforilação da enzima. Na sua forma activada a acetil-CoA
carboxilase polimeriza-se em longos filamentos.
Esta enzima é também regulada por modificações covalentes. A fosforilação,
provocada pelas hormonas epinefrina e glicagina, inactiva a enzima e reduz a sua capacidade
à activação pelo citrato, retardando assim a síntese de ácidos gordos; a fosforilação é
acompanhada pela sua dissociação em subunidades monoméricas e pela consequente perda de
actividade.
Nos vertebrados, o palmitoil-CoA, que é o produto principal da síntese de ácidos gordos, é um
inibidor retrógrado da enzima.
A enzima pode ainda ser inactivada por moléculas de acil-CoA de cadeia longa.
Se a síntese de ácidos gordos e a β-oxidação se dessem ao mesmo tempo, os 2
processos constituiriam um ciclo fútil, perda de energia. Assim durante a síntese de ácidos
gordos, a produção do primeiro intermediário, o malonil-CoA, não permite a β-oxidação ao
nível da membrana mitocondrial interna.
Quando uma célula ou organismo tem mais “combustível” metabólico do que o
necessário para as suas necessidades energéticas, este excesso é convertido em ácidos gordos
e armazenado como lípidos.
Há uma relação inversa entre lipogénese hepática e a concentração de ácidos gordos
livres. Quando ingerimos excesso de ácidos gordos insaturados a expressão dos genes que
codificam muitas enzimas lipogénicas no fígado é suprimida. Esta regulação da actividade enz
34
2.11- Síntese dos Lípidos
Biosíntese dos Triacilgliceróis
O triacilglicerol (TAG) é um éster derivado dos ácidos gordos e de um único álcool, o
glicerol. É assim formado pela união de três ácidos gordos a uma molécula de glicerol,
substituindo estes os três grupos hidroxilo (-OH) do glicerol pelos seus, formando moléculas
de água durante o processo. É normalmente identificado como um óleo ou gordura e é
produzido e armazenado nos organismos vivos para fins de reserva alimentar. Os
triacilgliceróis constituem cerca de 90% dos lípidos ingeridos na alimentação e no organismo
os seus locais de síntese incluem o retículo endoplasmático liso e algumas enzimas
localizadas no citosol e na mitocôndria. São compostos essencialmente apolares, pois as
regiões polares dos seus precursores desapareceram na formação das ligações do tipo éster.
Por isso constituem moléculas muito hidrofóbicas que são insolúveis em água e solúveis em
solventes orgânicos, como o álcool, benzeno, éter e clorofórmio. Os triacilgliceróis podem ser
hidrolisados, libertando com isso ácidos gordos e glicerol.
O principal precursor dos triacilgliceróis é o glicerol-3-fosfato. Este pode ser obtido
principalmente de duas formas: através da glicólise, a glicose sofre a acção da enzima
glicerol-3-fosfato desidrogenase citossólica que se encontra ligada ao NAD ou então através
da enzima glicerol cinase em processos que ocorrem no fígado e no rim, sendo que neste caso
o glicerol proveniente do metabolismo dos quilomicrons é transformado directamente em
glicerol-3-fosfato.
Numa primeira fase, são removidos os dois primeiros grupos hidroxilo livres do
glicerol-3-fosfato e adicionados dois ácidos gordos aos pontos respectivos de esterificação por
duas moléculas de acil CoA sintetase, sendo que esta reacção ocorre na presença da
aciltransferase existente na mitocondria. Desta primeira fase resulta a molécula de ácido
fosfatídico (dois ácido gordos e um grupo fostato ligados a uma molécula de glicerol). Neste
ponto a via pode seguir para a formação de triacilglicerol ou de glicerofosfolipídio, como
veremos adiante. Pela via do triacilglicerol existem mais 2 passos. No primeiro, o grupo
fosfato é hidrolisado pela fosfatidato fosfatase presente no citosol para formar um 1,2diacilglicerol. De seguida o diacilglicerol é convertido em triacilglicerol por transesterificação
com um terceiro acido gordo na presença da enzima aciltransferase.
Os triacilgliceróis seguem nessa altura 2 vias: aqueles que são ingeridos são
transportados para os tecidos através de lipoproteínas específicas, os quilomicrons, enquanto
que os formados no fígado pelo processo acima enunciado são transportados para os tecidos
extra-hepáticos (muscular e adiposo) pelas “very low density lipoproteins” (VLDL).
35
Biosíntese dos Fosfolípidos
A síntese dos fosfolípidos (que como já sabemos se dividem principalmente em
glicerofosfolípidos e esfingolípidos) ocorre principalmente no retículo endoplasmático liso nas células eucariotas - e pode-se dividir em 4 passos:
1-Síntese da molécula que vai ser a “espinha dorsal” do fosfolípido (glicerol ou
esfingosina)
2-Ligação de ácidos gordos a essa estrutura por ligação éster ou amida
3-Adição de uma cabeça hidrofílica através duma ligação fosfodiéster
4-Alteração ou troca do grupo hidrofílico para termos a estrutura final
(este último passo só acontece em alguns casos)
Nos glicerofosfolípidos os passos 1 e 2 são comuns à via dos triacilglicerois: dois
ácidos gordos são esterificados no C1 e C2 do L-glicerol-3-fosfato. Normalmente, mas não
necessariamente, existe um ácido saturado em C1 e um ácido insaturado em C2. O 3º passo –
a adição da cabeça hidrofílica – é o passo fulcral: é quando o diacilglicerol (o principal
percursor da síntese dos glicerofosfolípidos) se liga a cabeça hidrofílica. Os dois grupos
hidroxilo ao reagirem com o ácido fosfórico formam um éster.
Existem duas estratégias para formar essa ligação fosfodiéster. O CDP (citidina
difosfato) pode estar ligado ou ao diacilglicerol ou à cabeça hidrofílica. Os organismos
procariotas utilizam exclusivamente a primeira estratégia, enquanto que os organismos
eucariotas podem usar ambas
O primeiro passo da síntese da cardiolipina e da fosfatidiletanolamina em E. coli é um
exemplo da primeira estratégia para ligação entre a cabeça e o CDP-diacilglicerol: O ataque
nucleofílico pelos grupos hidroxilo da serina e do glicerol 3-fosfato originam respectivamente
a fosfatidilserina e o fosfatidilglicerol 3 fosfato (que só depois perderá esse grupo fosfato
formando-se o fosfatidilglicerol) A fosfatidilserina por descarboxilação transforma-se então
na fosfatidiletanolamina. Duas moléculas de fosfatidilglicerol podem-se juntar e, com a
eliminação de um glicerol, originar cardiolipina.
Caso o organismo em questão seja um eucariota a síntese da cardiolipina será
diferente: fosfatidilglicerol irá juntar-se ao CDP-diacilglicerol e não a outra molécula de
fosfatidilglicerol. Num organismo eucariota o fosfatidilinositol é sintetisado pela reacção do
CDP-diacilglicerol com o inositol. O Fosfatidilinositol poderá, por cinases específicas,
originar compostos derivados que serão muito importantes na transducção de sinal.
As leveduras podem produzir a fosfatidiletanolamina por descarboxilação da
fosfatidilserina. Importante é o facto da fosfatidilserina e fosfatidiletanolamina serem
interconvertíveis por uma reacção de troca de cabeças. Por metilação poderá ser obtida a
partir desta última a fosfatidilcolina
36
Por outro lado, nos mamíferos, o processo é inverso: a fosfatidilserina provem da
reacção de interconversão com a fosfatidiletanolamina, sendo que a síntese desta é um dos
exemplos da segunda estratégia anteriormente referida.
Nos mamíferos existe um processo de reutilização da colina, sendo esta convertida em
CDP-colina e produzindo-se depois a fosfatidilcolina. Por um processo análogo a etanolamina
é
recuperada
tranformando-se
em
CDP-etanolamina
e
posteriormente
em
fosfatidiletanolamina.
As células dos mamíferos têm processos semelhantes aos das bactérias excepto para
sintetizar a fosfatidiletanolamina e a fosfatidilcolina. Nestes organismos, esta conversão
apenas ocorre no fígado
A síntese dos plasmogénios envolve o PAF(platelet-activating factor) que é um
plasmogénio específico que funciona como mediador biológico poderoso. Esta síntese iniciase na dihidroxiacetona-fosfato, envolve a formação de uma ligação éter, seguida da ligação de
uma cabeça hidrofílica e por último a formação de uma ligação dupla por uma desaturase.
Quanto à síntese dos esfingolípidos esta ocorre em 4 fases:
1- Síntese da amina de 18 carbonos – esfinganina a partir da serina e do palmitil-coA
2- Ligação do ácido gordo por uma ligação de amida
3- Formação da ceramida a N-acil-esfingosina
4- Ligação da cabeça hidrofílica: um composto glicosídico ou uma fosfatidilcolina,
obtendo-se respectivamente um cerebrósido ou uma esfingomielina.
Os fosfolípidos após serem sintetizados no R.E.Liso vão ser transportados para as
membranas celulares específicas por vesículas de transporte e proteínas específicas, não sendo
transportados por difusão simples, porque não são hidrosolúveis.
Biossíntese do Colesterol e Esteróides em geral
O colesterol desempenha um papel crucial como componente das membranas celulares
e como percursor de hormonas esteróides e ácidos (sais) biliares. É uma molécula essencial
em muitos animais, incluindo os humanos, mas não é requerida na dieta dos mamíferos, pois
todas as células (mas principalmente as hepáticas) podem sintetizá-lo a partir de um simples
percursor: o acetato.
Este facto foi demonstrado por Konrad Bloch, em 1940, que ao marcar com isótopos
os carbonos do acetato (CH3-COO-) e fornecê-lo como alimento a ratos, verificou que o
colesterol (27 Carbonos) produzido possuía esses carbonos marcados.
Outra pista importante foi a descoberta do esqualeno (30 Carbonos) e das unidades
isoprenóides (cada uma com 5 carbonos).
37
Separemos então a síntese do colesterol em 4 passos que passo a explicar:
1 – Partindo de 3 moléculas de acetil-CoA, formamos o mevalonato (C6):
Duas moléculas de acetil-CoA, por acção de uma tiolase, formam acetoacetil-CoA,
que junto com mais um acetil-CoA, por acção da enzima HMG-CoA-sintase forma o
intermediário HMG-CoA (HMG-CoA = Hidroximetilglutaril-CoA). Finalmente, por acção da
HMG-CoA redutase, com 2NADPH + 2H+ , obtemos o mevalonato.
2 – O segundo passo consiste em converter o mevalonato nos isoprenos activos:
A partir do mevalonato, por acção da mevalonato 5-fosfotransferase, e
fosfomevalonato cinase, 3 grupos fosfato são transferidos de ATP para a molécula formando
os
intermediários:
5-fosfomevalonato,
5-pirofosfomevalonato
e
3-fosfo-5-
pirofosfomevalonato, que por sua vez sofre uma descarboxilação, e perdendo um fosfato,
surge o isopentenil-pirofosfato, que por uma isomerase, transforma-se também em dimetilalilpirofosfato (que são ambos os isoprenos activos).
3 – Condensação das seis unidades isoprenóides para formar o esqualeno:
Os dois isómeros mencionados anteriormente condensam-se “cabeça” com “cauda”, e
por libertação de um pirofosfato formam geranil-pirofosfato (C10). Este por sua vez,
condensa, também “cabeça” com “cauda” com um isopentenil-pirofosfato, libertando novo
pirofosfato e formando farnesil-pirofosfato (C15). Duas moléculas de farnesil-pirosfato
condensam, desta vez “cabeça” com “cabeça” formando assim, com libertação de ambos os
grupos pirofosfato, o esqualeno (C30 e linear).
4 – O último passo consiste na ciclização do esqualeno para formar o colesterol:
Na presença de oxigénio molecular (O2), vemos que a enzima esqualenomonoxigenase usa um desses átomos para formar o esqualeno-2,3-epóxido, enquanto o O
restante é reduzido pelo NADPH em H2O. Isto acontece num sistema complexo que envolve
por exemplo o citocromo P-450. Por acção de ciclases, forma-se o intermediário lanosterol,
que após uma série de reacções (principalmente migrações e remoções de grupos metilo) se
converte finalmente em colesterol. No caso das plantas, partindo do esqualeno-2,3-epóxido,
teríamos o estigmasterol e nos fungos, o ergosterol. De notar que partindo do esqualeno (C30)
se obtém o colesterol (C27), em que os carbonos perdidos se devem à formação dos anéis do
núcleo esterólico.
Para além do colesterol e seus derivados (hormonas esteróides, sais biliares e vitamina
D), a partir de intermediários como o isopentenil-pirofosfato conseguimos formar outros
compostos (vitaminas lipossolúveis: A,E,K; borracha natural, carotenóides, componentes da
clorofila, transportadores da cadeia electrónica como a ubiquinona e a plastoquinona, entre
outros).
38
Estudando as hormonas esteróides, conclui-se que a sua formação a partir do
colesterol deve-se por oxidação e clivagem das suas cadeias laterais, dependendo de um
sistema complexo, com citocromo P-450, adrenodoxinas, presença de NADPH e O2. São
efectivas em concentrações mínimas, e comparativamente aos sais biliares, consomem pouco
colesterol.
Forma-se primeiramente a pregnenolona, a partir da qual se formam as restantes
hormonas: no córtex das glândulas supra-renais – mineralocorticóides (controlo da reabsorção
de iões inorgânicos como Na+, Cl- e HCO3- pelos rins) e glicocorticóides (regulação da
gliconeogénese e redução da resposta inflamatória). E nas gónadas, a formação das hormonas
sexuais (progesterona (regulação do ciclo reprodutor feminino), androgénios e estrogénios
(regulação dos caracteres sexuais secundários masculino (testosterona) e feminino (estradiol),
respectivamente)).
O último tópico é então a formação dos sais biliares, pois o organismo só consegue
excretar o colesterol na sua forma original ou como ácidos biliares. Estes podem ser:
primários, se forem produzidos no fígado (como o ácido cólico e quenodesoxicólico); ou
secundários, se formados no intestino (caso do ácido desoxicólico e litocólico).
Por comparação das estruturas de sais biliares e colesterol, concluiu-se que para
formar os primeiros a partir do segundo ocorrem principalmente o desaparecimento da ligação
dupla, bem como hidroxilações sucessivas por monoxigenases (dependentes também do
citocromo P-450, NADPH e O2).
Regulação da biossíntese dos triacilglicéridos
Depois de clarificados os processos de biossíntese de triacilglicerois, fosfolípidos, e do
colesterol e seus derivados, importa perceber as vias, pelas quais estes processos vão ser
regulados, de forma proveitosa para o organismo.
Sendo os ácidos gordos produtos iniciais de várias vias metabólicas, é importante
perceber o que provoca o desencadeamento de algumas destas em detrimento das restantes;
assim, para uma determinada concentração de ácidos gordos, a formação preferencial de
reservas energéticas, na forma de triacilglicerois, ou a de lípidos de membranas vai depender
do estado de maturação do organismo em questão. Como exemplo, temos o caso de uma
célula em crescimento, que passa por processos de divisão celular, e na qual a concentração
de ácidos gordos vai ser preferencialmente usada para a formação de fosfolípidos, utilizados
como constituintes das membranas celulares em formação. Esta regulação processa-se
também a nível hormonal, assumindo a insulina, neste campo, um papel fundamental. Por
observação da figura seguinte, podemos verificar que esta intervenção se processa a 2 níveis.
39
Figura 1 – Regulaçao da biossíntese dos triacilglicerois pela insulina
Assim, a insulina estimula a biossíntese de triacilglicerois, quer pela estimulação da
glicólise, quer pela transformação do acetil-coA a ácidos gordos; daqui, é fácil perceber, que
em doenças que envolvam a desregulação funcional da insulina, como a Diabetes Mellitus, o
acetil-coA proveniente da glicólise vai ser preferencialmente usado em vias alternativas à
formação de triacilglicerois, como a formação de corpos cetónicos.
Depois da formação de ácidos gordos, entramos noutro nível de regulação: o ciclo
sistémico de regulação dos triacilglicerois, ilustrado na figura seguinte:
Figura 2 – Ciclo sistémico de regulação dos triacilglicerois
Sabe-se que 75% dos ácidos gordos libertados pela lipólise são re-esterificados a
triacilglicerois, no seguimento deste ciclo, em vez de serem utilizados como combustível.
Este facto mantém-se praticamente inalterado mesmo em condições de jejum. O
funcionamento deste ciclo, perante condições de necessidade energética, consiste na
libertação de ácidos gordos do tecido adiposo para a corrente sanguínea, onde 25% dos
mesmos, serão utilizados em processos oxidativos para a produção de energia. A nível
40
hormonal, esta libertação de ácidos gordos é estimulada pela glicagina e pela epinefrina. A
utilidade deste ciclo é questionável, existindo no entanto uma vantagem evidente que reside
no facto, deste constituir uma reserva de energia que permite uma resposta muito rápida. O
facto da razão de re-esterificação de ácidos gordos se manter aproximadamente constante,
mesmo em jejum, leva à necessidade de existência de uma via que permita manter a
concentração de glicerol- 3P constante. Esta via é a gliceroneogénese, que permite a
transformação do piruvato a dihidroxiacetona (DHAP), que será posteriormente transformada
a glicerol-3P; a sua importância reside no controlo da taxa de ácidos gordos libertados para o
sangue. A gliceroneogénese é controlada enzimaticamente pela expressão da PEPCK (PEP
carbocinase) que estimula esta via metabólica. A regulação transcripcional da PEPCK é feita
por enzimas glicocorticóides (cortisol e dexametasona). Como pode ser observado na figura
??, estas enzimas actuam de forma antagonista no fígado e tecido adiposo; assim, as
glicocorticoides vão activar a PEPCK no fígado, e como tal, estimular a síntese e exportação
de triacilglicerois, e inibir a expressão da PEPCK no tecido adiposo diminuindo a reciclagem
de ácidos gordos e, assim aumentar a sua concentração no sangue.
Sendo a biossíntese do colesterol um processo complexo e que envolve o gasto de
energia (ATP), é vantajoso regular este processo em conjunto com a ingestão diária. Como foi
referido atrás, o passo limitante na via do colesterol é a conversão do HMG-coA a
mevalonato; esta reacção é catalisada pela HMG-coA reductase, que é regulada
transcripcionalmente e a nível hormonal. Ao nível da transcrição, o gene que a codifica é
regulado pela família de proteínas SREBPs (sterol binding element-binding proteins). Estas,
aquando da sua síntese, localizam-se no retículo endoplasmático (RE). Sendo que, apenas o
terminal amina funciona como activador transcripcional, logo a activação do gene da HMGcoA reductase só ocorre após a clivagem proteolítica e posterior migração para o núcleo, do
terminal amina. Perante níveis elevados de colesterol, a SREBPs permanecem inactivas
formando um complexo com a SCAP no RE. Quando os níveis de colesterol baixam vai haver
libertação deste complexo e posterior dupla clivagem proteolítica que permite a activação do
gene, e posterior produção de colesterol. A nível hormonal, vamos ter um controlo
proporcionado pela insulina que favorece a forma activa da enzima através duma
desfosforilação, e pela glicagina que através duma fosforilação produz a forma inactiva.
Existem ainda outros mecanismos: elevados níveis de colesterol intracelular activam a ACAT
que promove a transformação do colesterol em ésteres de colesterol, e por fim, elevados
níveis de colesterol extracelular diminuem a transcrição do gene codificador dos receptores de
LDL, que, por endocitose promovem o uptake de colesterol para dentro da célula.
41
3.1- Catabolismo e Biossíntese de ácidos nucleicos,
nucleótidos e nucleósidos
Os ácidos nucleicos são constituídos por nucleótidos que são compostos, por sua vez,
por uma base azotada, uma pentose e um grupo fosfato.
Existem bases púricas e pirimídicas, que são derivadas da purina e da pirimidina
respectivamente.
As bases púricas são a adenina ( 6–aminopurina ) , a guanina ( 2–amino–6–oxipurina )
, a hipoxantina ( 6-oxipurina ) e a xantina ( 2,6-dioxipurina ). As bases pirimídicas são a
citosina ( 4-amino-2-oxipirimidina ), o uracilo ( 2,4-dioxipirimidina ), a timina ( 5-metil-2,4dioxipirimidina ), o ácido orótico ( 2,4-dioxi-6-carboxipirimidina ).
As nucleoproteínas que são ingeridas na dieta sofrem a acção de proteases e são
degradadas em proteínas e ácidos nucleicos. Os últimos são transformados em nucleótidos
pela acção de nucleases.
Existem dois tipos de nucleases:
-
Endonucleases: cindem ligações no interior da cadeia polinucleotídica e podem originar
nucleótidos 5’-fosfoterminais ( por exemplo, as desoxirribonucleases no pâncreas, por
cisão de ligações 3’-5’ ) ou nucleótidos 3’-fosfoterminais ( por exemplo, as
desoxirribonucleases no baço, por cisão de ligações entre um grupo fosfato e o carbono 5’
);
-
Exonucleases: cindem ligações fosfodiéster nas extremidades da cadeia polinucleotídica.
Os nucleótidos são então desfosforilados por hidrólise em nucleósidos ( por acção de
nucleotidases ).
Os nucleósidos púricos ( adenosina e guanosina ) sofrem catabolismos diferentes:
-
A adenosina é desaminada por acção da adenosina desaminase formando-se inosina. Em
seguida, esta sofre a acção de uma enzima – a nucleosidase – perdendo a sua pentose (
ribose ou desoxirribose ) e origina-se hipoxantina, a qual é oxidada pela acção da xantina
oxidase ( o aceitador de electrões é o oxigénio molecular ) formando-se xantina.
-
A guanosina perde a sua pentose através da actuação de uma nucleosidase, formando-se
guanina. Esta é desaminada ( por acção da guanina desaminase ) ocorrendo formação da
xantina.
-
A xantina é um composto comum à degradação da adenosina e guanosina e é oxidada a
ácido úrico por acção da xantina oxidase.
42
O produto final do catabolismo de purinas é o ácido úrico, que é o produto de excreção
dos seres humanos.
Os nucleósidos pirimídicos ( citidina, uridina e timidina ) são degradados a ureia,
ácido propiónico, acético, amoníaco e dióxido de carbono da seguinte forma:
-
O nucleósido é hidrolisado por acção de uma nucleosidase, perdendo a sua pentose e
originando a base pirimídica correspondente;
-
A citosina pode ser desaminada e originar uracilo ou então pode ser metilada, formandose metil-citosina, a qual é desaminada e dá origem à timina;
-
O uracilo é reduzido, por oxidação do NADPH e forma-se di-hidrouracilo, sendo este
transformado em ureia e
-
-alanina ( sendo esta degradada a CO2, NH3 e ácido acético );
A timina é igualmente reduzida por oxidação do NADPH, formando-se di-hidrotimina,
-aminoisobutirato ( que é transformado em CO2 e ácido propiónico ).
Resumidamente:
-
Nas purinas o anel púrico é mantido no produto final do seu catabolismo, o ácido
úrico.
-
Na pirimidinas o anel pirimídico é destruído, daí o aparecimento de aminoácidos e
outros produtos.
Biossíntese das purinas e derivados:
Os nucleótidos contêm resíduos de ácido fosfórico, de um açúcar (em geral uma pentose) e de
uma base púrica (ou pirimídica). Quer as bases púricas, quer as pirimídica são anéis
heterocíclicos contendo átomos de azoto e carbono. As bases púricas podem ser entendidas
como constituídas por um anel de pirimidina (anel com seis átomos, quatro de carbono e dois
de azoto) ligado a um anel de imidazol (anel com cinco átomos, três de carbono e dois de
azoto). São bases púricas a adenina, a guanina, a hipoxantina e a xantina.
Por hidrólise dos nucleótidos púricos, geram-se os respectivos nucleósidos . Estes resultam da
união entre uma purina e um açúcar em que a ligação envolve o átomo N9 da base.
Existem dois tipos de via que levam à síntese de nucleótidos. São elas a via “De novo” e via
de reaproveitamento ou “salvage pathway”. Esta segunda via, cujas reacções são catalisadas
pelas transferases de fosforibosilo e pelas cinases de nucleósidos, permite recuperar bases e
nucleósidos a nucleótidos. O anel de purina é construído passo a passo. Este processo começa
com a 5-fosforibosilamina. Os aminoácidos glutamina, glicina e aspartato, fornecem todos os
átomos de azoto do anel da purina.
43
Na síntese de novo das purinas os intermediários contêm ribose-5’-fosfato e o primeiro
nucleótido formado é a inosina-5’-fosfato (IMP) cuja base é a hipoxantina (6-oxipurina). Na
primeira reacção a ribose-5-P, formada na via das fosfopentoses, funciona como aceitador dos
grupos fosfato - do ATP que se ligam no carbono 1 da ribose gerando-se PRPP; esta reacção
é catalisada pela PRPP síntase. Na segunda reacção ocorre ruptura da ligação formada na
primeira reacção e transferência do azoto do grupo amida da glutamina para o carbono 1 da
ribose formando-se 5-fosforibosilamina; é catalisada pela amido-fosforibosil-transferase da
glutamina. O azoto N9 do anel purina deriva assim da glutamina. Sucessivamente vão-se
incorporando a glicina (C4, C5 e N7), uma unidade monocarbonada cedida pelo N10-formilH4-folato (C8), outro azoto do grupo amida da glutamina (N3), o CO2 (C6), o grupo amina do
aspartato (N1) e outra unidade monocarbonada cedida pelo N10-formil-H4-folato (C2). O
processo mostra que a formação de alguns dos intermediários fosforibosilo da via metabólica
está acoplada com a rotura de ligações fosfoanidrido do ATP.
É a partir do IMP que se formam quer o AMP (por aminação do carbono 6) quer o GMP
(oxidação dependente do NAD+ e posterior aminação do carbono 2). O dador do grupo 6amina do AMP é o aspartato; na formação do AMP a partir do IMP intervém a sintetase de
adenilosuccinato (IMP + aspartato + GTP
adenilosuccinato + GDP + Pi). O dador do
grupo 2-amina do GMP é a glutamina; na formação do GMP a partir do IMP intervém uma
desidrogenase (desidrogenase do IMP: IMP + NAD+
XMP + NADH) que origina XMP,
o intermediário que funciona como aceitador do grupo amina da glutamina. A regulação deste
processo é feita por feedback negativo. Assim, a transformação de IMP em AMP requer GTP
e a transformação de IMP em GMP requer ATP, o que permite o estabelecimento de um
equilíbrio. De qualquer forma, quando há AMP ou GMP em excesso, há uma inibição parcial
do processo e não total (excesso de GMP nao afecta formação de AMP).
Os nucleósidos monofosfato são convertidos em nucleósidos trifosfato por reacções de
fosforilação enzimática. A fosforilação de AMP a ADP é promovida pela adenilato cinase. O
ADP formado é posteriormente fosforilado a ATP por fosforilação oxidativa ou por enzimas
glicolíticas.
Na via de reaproveitamento (“salvage pathway”), as bases púricas livres, são reutilizadas para
formar novos nucleótidos. A base (adenina livre) liga-se ao PRPP (1.) por acção da PRPP
transferase e ocorre combinação do composto formado em 1. com a fosforibose para dar
origem ao nucleósido que é fosforilado.
Adenina + PRPP AMP + PPi
Os ribonucleótidos são precursores dos desoxirribonucleótidos. Estes últimos derivam
directamente dos ribonucleótidos correspondentes por redução directa no átomo de carbono
C2 da ribose.
44
3.2- Síntese de aminoácidos não essenciais
Glutamato
O glutamato é formado nas mitocôndrias dos hepatócitos, através de uma transaminação – o α
amino grupo de um aminoácido é transferido para o carbono α do cetoglutarato.
É dador do ião amónio dentro da célula.
Glutamina
A glutamina é formada, normalmente, quando há excesso de NH4+ nos tecidos. È um
transportador não tóxico deste ião e forma-se nos rins, fígado e intestino, sendo também uma
fonte de grupos amina em várias vias.
A glutamina é formada a partir do glutamato que reage com ATP, formando-se
glutamilfosfato, que ao perder um fosfato inorgânico e
com a adição de mais um grupo amina origina então a
glutamina. Estas duas reacções são sintetizadas pela
enzima glutamina sintetase.
No fígado e nos rins existe uma enzima, a glutaminase,
que permite a formação de glutamato a partir da
glutamina.
Prolina
A prolina também se forma a partir do glutamato.
1 - O carbono γ do glutamato reage com o ATP
originando o γ – glutamilfosfato. A reacção é catalisada
pela enzima glutamato-cinase.
2 – O γ – glutamilfosfato, através da enzima glutamato
desidrogenase é reduzido pelo NAD(P)H, originando,
após a perda de um fosfato inorgânico, o glutamato γ –
semialdeído. Esta reacção é catalisada pela enzima
glutamato desidrogenase.
3 - glutamato γ – semialdeido, por um processo rápido e
não enzimático, sofre uma ciclização originando o P5C,
que após redução por uma molécula de NAD(P)H, origina
a prolina.
Serina
A serina é formada a partir do 3 – fosfoglicerato, um composto
intermediário da via da glicólise.
1 – O grupo hidroxilo do 3 – fosfoglicerato, é oxidado pela molécula de
NAD+, através da fosfoglicerato desidrogenase, originando 3 –
fosfohidroxipiruvato.
2 – Através de uma transaminação o glutamato perde o grupo amina,
originando a 3 – fosfoserina que é hidrolisada pela fosfoserina fosfatase,
originando-se serina.
3 – A serina é o percursor da glicina. A serina perde um carbono, através
da enzima hidroximetiltransferase, que é aceite pelo tetrahidrofolato,
originando-se glicina.
45
Cisteína
Os mamíferos sintetizam cisteína através de dois aminoácidos: a
metionina, que fornece o átomo de enxofre, e a serina que fornece
o esqueleto de carbonos.
A metionina é convertida em homocisteína que reage com a
serina, formando-se a cistationina γ, através da enzima
cistationina βsintetase. A cistationina, origina a cisteína e o α –
cetobutiraro.
A enzima cistationina γ liase, interfere nesta última reacção, e
necessita de um cofactor, o PLP (fosfato peridoxal, um cofactor
que interfere em descarboxilações, transaminações e
desaminações).
Aspartato:
O aspartato é sintetizado através do oxaloacetato
(intermediário do ciclio de Krebs) com uma transaminação
do glutamato. Ocorre no fígado, rins e coração.
Asparagina:
A asparagina é sintetizada pela amidação do aspartato com
a glutamina a doar NH4+.
Alanina:
A alanina é sintetizada através do
piruvato por transaminação do glutamato.
Tirosina
A tirosina é obtida através da fenilalanina com a hidroxilação do
carbono quatro do grupo fenilo, pela enzima fenilalanina
hidroxilase.
Hidroxiprolina e Hidrolisina
Hidroxiprolina e Hidrolisina mantêm a estrutura tridimensional do colagénio, principalmente
logo após a sua formação.
A hidroxiprolina é formada através da prolina com a junção de um grupo hidroxilo. (reacção
catalisada pela Prolil-4-hidroxilase).
A hidroxilisina é formada através da lisina com a junção de um grupo hidroxilo. (reacção
catalisada pela Lisil-4-hidroxilase).
Produtos derivados de alguns aminoácidos
Serina
Selenocisteína – Ao contrário do que o nome possa sugerir, é formada a
partir da serina. Contém um átomo de selénio (ligado a outro de hidrogénio)
no lugar onde estaria o grupo hidroxilo na serina.
Fosfoserina – Em reacções que ocorrem após a tradução, a serina (na forma
de resíduo de aminoácido) pode ser fosforilada por diversas cinases. A fosforilação ocorre no
46
grupo hidroxilo e constitui um ponto de regulação para diversos mensageiros celulares e
enzimas.
Metionina – É um precursor da homocisteina,
referida anteriormente na síntese da cisteína.
Inicialmente combina-se com o ATP formando
S-adenosilmetionina. Sob esta forma perde um
grupo metilo, formando-se S-adenosilhomocisteina. A ligação entre a adenosina e a
homocisteina é então hidrolisada, formando-se
assim os compostos finais.
Importância do ácido fólico
O ácido fólico tem a sua actividade sob a forma
reduzida de tetrahidrofolato, este funciona como
dador de carbono. O carbono pode ser obtido pela
formação da glicina a partir da serina ou por adição
directa de formato (CHOO-), sendo para esta última
reacção necessário o gasto de uma molécula de
ATP. O carbono pode ser cedido sob a forma de
diversos grupos funcionais. Esses grupos por ordem
crescente de estado de oxidação são: metilo (-CH3),
hidroxilo (-CH2OH), forminino (-C=NH) e
carboxilo (-CHO).Um exemplo de uma reacção em
que o tetrahidrofolato participa é a metilação da
homocisteina a metionina.
Tirosina – É um percursor da dopamina, norepinefrina e epinefrina. Para a
produção de dopamina é adicionado um grupo hidroxilo ao anel fenilo da
tirosina, neste passo a tetrahidrobiopterina actua como cofactor da tirosina
hidroxilase. Forma-se assim dihidroxifenilalanina (dopa). A dopamina surge por
descarboxilação da dopa, catalisada por uma descarboxilase dos AA aromáticos.
A norepinefrina surge por meio de uma nova adição de um grupo hidroxilo,
desta vez ao carbono β da dopamina, através da dopamina β-hidroxilase. A
epinefrina obtém-se por metilação do grupo amina da norepinefrina através da
feniletanolamina N-metiltransferase.
A dopamina e a norepinefrina são neurotransmissores e a epinefrina é uma
hormona. Estes produtos são denominados por catecolaminas.
Treonina – Tal como a serina, a treonina pode também ser fosforilada sob a
forma de resíduo de aminoácido, constituindo um importante ponto de regulação
para diversas proteínas. Neste caso a fosforilação ocorre com a substituição do
grupo hidroxilo.
47
Arginina – A arginina é utilizada como fonte de um átomo de azoto para a sintese do
monóxido de azoto, um importante vasodilatador, que exerce a sua actividade relaxando os
endotélios dos vasos sanguíneos. A arginina reage
com o oxigénio e é utilizado NADPH como
redutor, numa reacção catalisada pela monoxido de
azoto sintase. Como produtos obtêm-se o
monóxido de azoto e citrulina, que terá como
destino o ciclo da ureia.
Ornitina – A ornitina pode ser utilizada como uma outra via para a
obtenção da prolina. Num primeiro passo, por acção de
uma δ-aminotransferase, o grupo amina do radical da
ornitina é transferido para o α-cetoglutarato. Forma-se
glutamato-γ-semialdeido, que sofre uma ciclização não
enzimática e, por uma reacção de redução (NAD(P)H é oxidado)
catalisada pela pirrolina carboxilato redutase, forma-se então a prolina.
Triptofano – Por adição de um grupo hidroxilo ao anel fenilo, através da
triptofano hidroxilase, e por posterior descarboxilação, por accção da
descarboxilase dos aminoáciso aromáticos, o triptofano dá origem à
serotonina, um neurotransmissor com propriedades antidepressivas.
Cisteína - Num passo pós tradução, a cisteína,
sob a forma de resíduo de aminoácido, pode
reagir com outro resíduo homólogo, formando uma ponte dissulfito
entre as respectivas cadeias polipeptídicas. Os dois resíduos de
cisteína, ligados pela ponte dissulfito denominam-se por cistina.
Estas podem conferir uma grande rigidez às proteínas em que estão
integradas, sendo responsáveis pela grande resistência mecânica de proteínas como a
queratina presente, por exemplo, nas unhas e cabelo humanos.
Formação de produtos de aminoácidos
Creatina
Na formação da creatina estão envolvidas a glicina e a
arginina. A arginina (por acção de uma amidinotransferase)
transfere um grupo amida para a glicina dando origem ao
guanidinoacetato. A creatina é o produto da metilação do
guanidinoacetato, através de uma metiltransferase, que
transforma a adenosilmenitionina (dador do grupo metilo)
em adenosil-homocisteína.
A fosfocreatina é usada para armazenar energia. O fosfato do
ATP é transferido para a creatina, formando a fosfocreatina
através da acção da fosfocinase. A reacção é reversível e o seu sentido depende das
necessidades energéticas da célula, sendo que o
equilíbrio está fortemente deslocado no sentido da
formação da fosfocreatina. Tanto a creatina como
a fosfocreatina podem ser encontradas no
músculo, cérebro e sangue.
48
Creatinina
A creatinina é formada no músculo a partir da fosfocreatina por uma desidratação não
enzimática e perda de um fosfato inorgânico. A creatinina é excretada pelos rins e a
depuração é uma medida da função renal.
Gama-aminobutirato (GABA)
O GABA é um neurotransmissor inibitório. É formado por
descarboxilação do glutamato através da acção de uma
glutamato descarboxilase.
Glutatião
O glutatião é formado a partir de três aminoácidos: glutamato,
cisteína e glicina. Inicialmente o glutamato liga-se à cisteína
(o grupo γ-carboxílico do glutamato é activado pelo ATP
formando acilfosfato que é depois “atacado” pelo grupo αamina da cisteína), por acção da γ-glutamil cisteína sintetase,
formando o γ-Glu-Cis. Seguidamente por acção da glutatião
sintetase a glicina é ligada aos dois péptidos iniciais (o grupo
α-carboxílico da cisteína é activado pelo ATP permitindo a ligação com a glicina).
O glutatião funciona como anti-oxidante (particularmente importante no ambiente oxidante
dos eritrócitos), está envolvido no transporte de a.a através das membranas celulares, serve
como cofactor em algumas reacções enzimáticas e participa no rearranjo das ligações
dissulfito nas proteínas.
A forma oxidada do glutatião consiste em duas moléculas do mesmo ligadas por uma ligação
dissulfito. A glutatião reductase utiliza NADPH como cofactor para reduzir o glutatião.
Poliaminas
Na síntese de poliaminas estão envolvidas, entre outros, a putrescina (catabolito da ornitina) e
a S-adenosil metionina, como dador de dois resíduos de propilamina. A função da ornitina
descarboxilase é produzir putrescina (4 carbonos) a partir de ornitina (5 carbonos), enquanto a
S-adenosil metionina descarboxilase (cuja actividade é estimulada pela putrescina,) “retira” o
grupo carboxilo da S-adenosil metionina.
A adenosil metionina descarboxilada juntamente com a putrescina e por acção de propilaminotransferase I dão origem à
espermidina
e
a
uma
metiltioadenosina. Um segundo
resíduo propilamina é adicionado à
espermidina por acção da propilaminotransferase II dando origem à
espermina
e
a
outra
metiltioadenosina.
As poliaminas são altamente
catiónicas e tendem a ligar-se aos
ácidos nucleicos. Com base neste
facto acredita-se que as poliaminas
participem na síntese de DNA, ou
na regulação desse processo.
49
3.3- Síntese e degradação das porfirinas e heme,
mecanismos reguladores e derivados
(a) Biossíntese
As porfirinas são uma classe de moléculas orgânicas derivadas de um macrociclo formado
por quatro anéis pirrol (C4H5N - aromático) ligados por ligações meténicas (=CH-). São
pigmentos de cor púrpura, devido ao carácter aromático do ciclo.
Este macrociclo, tendo os 4 átomos N voltados para o interior, possui no seu centro um
espaço apropriado para acomodar um ião metálico (estabilizando-o por ressonância).
Designam-se por porfirinas livres ou ligadas caso possuam ou não este ião. Exemplos de
porfirinas ligadas incluem o grupo heme (Fe2+) e a clorofila (Mg2+). Estes compostos
comportam-se frequentemente como compostos de coordenação, em que o ião metálico pode
ter capacidade de ligar mais um ou dois grupos químicos no eixo perpendicular ao plano do
anel da porfirina. No caso do grupo prostético heme, a função a desempenhar varia com a
proteína que o contém:
•
Hemoglobina, Mioglobina - ligação de O2 ou CO ao ião Fe2+.
•
Catalase – alteração do estado de oxidação do ião Fe3+, catalisando a dismutação do
peróxido de hidrogénio.
•
Citocromos- transporte electrónico através do ião Fe3+/Fe2+.
A biossíntese das porfirinas em humanos ocorre principalmente nas células hepáticas e da
medula óssea, embora possa ocorrer em todas as células do organismo, e tem início com a
formação de δ– aminolevulinato (δ -ALA). Na mitocôndria, o aminoácido glicina
(proveniente do citosol) reage com succinil- CoA (intermediário do ciclo de Krebs) formando
α- amino- β- cetoadipato, sendo este descarboxilado a δ-ALA. As 2 reacções são catalisadas
pela ALA (aminolevulinic acid) sintase.
As moléculas de δ-ALA juntam-se 2 a 2, (enzima: porfobilinogénio sintase/ ALA
desidratase), saindo da mitocôndria e originando por desidratação moléculas de
porfobilinogénio, um derivado do anel pirrol. Por cada 4 destas moléculas forma-se, através
de desaminação, (enzima: porfobilinogénio desaminase/ uroporfirinogénio sintase) uma
molécula de pré-uroporfirinogénio, sendo esta desidratada (enzima: uroporfirinogénio III
cosintase) a uroporfirinogénio III, a primeira porfirina. De seguida ocorre descarboxilação dos
4 substituintes acetilo da molécula anterior (enzima: uroporfirinogénio III descarboxilase),
formando-se coproporfirinogénio III. O mesmo entrará de novo na mitocôndria, originando
protoporfirinogénio (enzima: coproporfirinogénio oxidase) por descarboxilação/oxidação de
dois dos 4 substituintes propionilo do macrociclo (tornam-se vinilo). A oxidação de 2 átomos
50
N do macrociclo (enzima: protoporfininogénio oxidase) dá origem à molécula de
protoporfirina, que por acção da enzima ferroquelatase vai incorporar no centro do macrociclo
um ião Fe2+ vindo do citosol. Temos então a molécula heme, produto final da via de síntese,
pronta a sair da mitocôndria a fim de se ligar a cadeias polipeptídicas (ex: cadeias de globina)
formando hemoproteínas. Em humanos, defeitos genéticos podem causar a desregulação deste
sistema por deficiência de uma das enzimas da via de síntese de porfirinas, causando a
acumulação de um dos intermediários. As doenças deste grupo (uma para cada enzima) são
conhecidas por porfírias. Em eucariotas superiores, o principal regulador da via é a
concentração de heme na célula, actuando por um mecanismo de regulação retrógrada. A
concentração de Fe2+, por outro lado, actua por retroacção positiva na transcrição da enzima
ALA sintase, 1ª enzima. No caso dos hepatócitos, em que a síntese de heme ocorre para
incorporação em citocromos (P450, c, entre outros), a hemina (heme na forma oxidada –
Fe3+, constituinte dos citocromos que participam na fosforilação oxidativa) inibe a enzima
ALA sintase, bem como a sua síntese e transporte para a mitocôndria. Nos precursores dos
eritrócitos, o objectivo da biossíntese de heme é a formação de hemoglobina, pelo que apenas
se dá aquando da diferenciação e maturação celular, período em que é sintetizada a
hemoglobina. Neste período, a concentração de heme estimula também a síntese proteica.
(b) Degradação e pigmentos biliares
Degradação das porfirinas
No ser humano, a maioria das porfirinas é utilizada sob a forma de grupos heme em
moléculas como a hemoglobina, a mioglobina, a catalase e os citocromos. A degradação
desses grupos heme, principalmente da hemoglobina, envolve processos que decorrem
principalmente no baço, na medula óssea e também no fígado. Nestes órgãos, os eritrócitos
danificados ou em final de vida são destruídos, libertando as suas moléculas de hemoglobina,
que são rapidamente fagocitadas por macrófagos (ou pelas células de Kupfer, no fígado). No
seu interior, as componentes proteicas (globina) são decompostas em aminoácidos, e os
grupos heme podem então ser degradados para fornecer iões Fe3+ e, posteriormente,
bilirrubina. Isto pode ser feito pelos macrófagos em todo o tipo de tecidos, através de uma via
metabólica de dois passos: no primeiro, catalisado pela enzima heme oxigenase, dá-se a
conversão de um grupo heme em biliverdina, um derivado do tetrapirrol de cadeia linear
(ocorrendo portanto a clivagem do anel porfírico). Obtêm-se também como produtos um ião
Fe2+ livre e uma molécula de CO, sendo esta a única reacção conhecida que produz CO no
organismo humano. O ferro, por um lado, é rapidamente captado pela transferrina, para
reaproveitamento. O CO irá ligar-se à hemoglobina para eventualmente ser expulso nos
51
pulmões. O segundo passo da via consiste na conversão da biliverdina em bilirrubina,
catalisada pela enzima biliverdina redutase (que utiliza uma molécula de NADPH como fonte
de poder redutor). A molécula de bilirrubina é também uma cadeia aberta de quatro grupos
pirrol, cuja conformação se pode alterar na presença de luz.
Pigmentos biliares
Tal como as moléculas de porfirina propriamente ditas, a biliverdina e a bilirrubina são
compostos com propriedades ópticas particulares, nomeadamente cores características. Isto
pode ser observado facilmente numa simples contusão: a danificação dos capilares provoca
derrame e coagulação de sangue, e consequente degradação local de eritrócitos, que libertam
hemoglobina. Com o tempo, a zona afectada, inicialmente negra ou roxa, passa a apresentar
uma cor esverdeada (devido à produção de biliverdina) e posteriormente amarelada (devido à
sua conversão em bilirrubina). Estes dois compostos são designados por pigmentos biliares.
A bilirrubina é muito pouco solúvel, pelo que só entra na corrente sanguínea depois de
associada à albumina sérica (ainda na forma não-conjugada). Uma vez chegada ao fígado,
grande parte da bilirrubina recém-formada entra nos hepatócitos e, no seu interior, desliga-se
da albumina e associa-se a complexos intermédios, que a transportam para o retículo
endoplasmático liso. Aqui, por acção da enzima glicuronil-bilirrubina transferase, a
bilirrubina é convertida principalmente em bilirrubina diglicuronato, um produto já
suficientemente solúvel para ser integrado na bílis e secretado para o intestino delgado.
Chegada ao intestino, a forma conjugada da bilirrubina pode ser convertida, por intermédio de
enzimas bacterianas, em vários outros compostos, principalmente urobilinogénio. Uma parte
deste produto (altamente solúvel) é reabsorvida para a circulação, sendo a maior parte captada
e reexcretada pelo fígado para o intestino. No entanto, uma fracção de cerca de 5% é
transportada para os rins, onde é oxidada em urobilina (que dá à urina a sua cor amarelada) e
excretada. Por outro lado, o urobilinogénio que permanece no intestino pode ser convertido
em estercobilina (que por sua vez confere às fezes a sua cor castanho-avermelhada) e
prosseguir ao longo do tracto intestinal. Todos estes produtos derivados da bilirrubina são
também considerados pigmentos biliares.
Efeitos nocivos dos pigmentos biliares
Problemas no funcionamento do fígado ou na secreção da bílis podem provocar fugas de
bilirrubina para a corrente sanguínea, resultando numa condição conhecida por icterícia, que
se caracteriza por uma amarelamento da pele, olhos e mucosas devido ao aumento acentuado
da concentração de bilirrubina no sangue (hiperbilirrubinemia). Isto também ocorre por vezes
em recém-nascidos que ainda não possuem níveis suficientes da enzima glicuronil-bilirrubina
52
transferase, necessária para o processamento da bilirrubina no fígado. Em condições normais,
é pouco prejudicial e desaparece com o ajustamento fisiológico pós-natal, ainda que, em
condições de maior fragilidade, possa verificar-se a acumulação excessiva e prejudicial do
pigmento em certas regiões cerebrais.
Um método de tratamento tradicional envolve a exposição a lâmpadas fluorescentes
(fototerapia), que induzem reacções fotoquímicas que levam à conversão da bilirrubina em
compostos mais solúveis e mais fáceis de excretar.
Importância da degradação das porfirinas
Os produtos da degradação dos grupos heme têm um papel bastante importante na regulação
de certas funções celulares e na protecção das células contra danos oxidativos. O CO
produzido pela heme oxigenase é de facto tóxico quando em concentrações elevadas, mas
quando produzido em baixos níveis, como na degradação dos grupos heme, parece
desempenhar certas funções de regulação e sinalização, e actuar como vasodilatador ou como
regulador de processos de neurotransmissão.
A bilirrubina, por outro lado, é o antioxidante mais abundante nos tecidos dos mamíferos,
actuando na protecção contra os danos oxidativos provocados por radicais livres e formas
activas de oxigénio (ROS), principalmente nos lípidos e proteínas membranares. Quando é
oxidada, passa a biliverdina, mas é rapidamente reconvertida em bilirrubina pela enzima
biliverdina redutase. Essa rapidez torna-a um antioxidante bastante poderoso, e de facto mais
eficaz que o glutatião (que requer a interacção com duas enzimas diferentes para ser oxidada e
novamente reduzida).
Regulação da degradação das porfirinas
Dada a grande variedade de funções dos produtos resultantes da degradação dos grupos
heme, esta via está sujeita a mecanismos de regulação bastante específicos, principalmente ao
nível do primeiro passo da degradação. Existem, no ser humano, pelo menos 3 tipos de
isozimas da heme oxigenase. A HO-1 é altamente regulada, sendo a sua produção induzida
por vários factores típicos de ambientes de stress celular. A HO-2, por outro lado, encontra-se
principalmente no cérebro e testículos, onde é continuamente produzida em condições
homeostáticas. A HO-3 não se encontra ainda bem caracterizada.
53
(c) Composição e mecanismo de acção do citocromo P450 na destoxificação
hepática
Importância Biomédica
Muitas drogas, poluentes e carcinogénios químicos (xenobióticos) são metabolizados
por oxigenases (incorporam oxigénio numa série de substratos, através de uma reacção em
que o oxigénio se liga à enzima no sitio activo, e posteriormente reduzido), conhecidas por
citocromo P450.
O nome de citocromo P450 vem do seu pico de absorção de aos 450 nm. È um dos biocatalisadores mais versáteis conhecidos pelo homem. As diferentes isoformas do citocromo
P450 constituem uma família de enzimas contendo Heme.
1) Dado o grande número de isómeros (cerca de 150), houve a necessidade de criar uma
nomenclatura sistemática:
CYP 1A1 (primeiro número família (40% de semelhança estrutural), letra subfamilia
(55% de semelhança estrutural), segundo número identifica dentro da família)
2) Tal como a hemoglobina, são hemeproteínas
3) Encontram-se presentes em maior quantidade no fígado e no intestino delgado. Existe
uma elevada concentração no retículo endoplasmático liso.
A estrutura do citocromo P450 é semelhante à de outros citocromos, apresentando cadeias
proteicas, e a presença de um grupo Heme.
As monooxigenases (oxidases de função mista) incorporam somente um átomo de oxigénio
molecular no substrato. O outro átomo de oxigénio é reduzido a água. Para tal, é necessário
um doador adicional de electrões, ou um co-substrato.
Formula geral da reacção:
A-H+O2+ZH2 -> A-OH + H2O + Z
Estes sistemas de monooxigenases microssomais e citocromo P450 são importantes para a
hidroxilação de muitas drogas, e actuam ao nível do fígado, juntamente com o citocromo b5.
O NADH e o NADPH participam na redução destes citocromos, os quais por sua vez são
oxidados pelos substratos numa série de reacções enzimáticas conhecido como ciclo das
hidroxilases.
54
DROGA-H + O2 + 2Fe2+ (citocromo P450) + 2H+ DROGA-OH + H2O + 2Fe3+ (citocromo
P450)
Entre os diferentes tipos de droga encontram-se o benzopireno, aminopirina, anilina,
morfina, benzoafetamina.
Os citocromos P450 também estão relacionados com a biossintese de hormonas esteróides,
mas essa função foge do âmbito do nosso trabalho.
Metabolismo de xenobióticos
Xenobiótico: Composto químico estranho ao organismo. Para serem excretados, têm de sofrer
alterações químicas, que ocorrem principalmente ao nível do fígado.
Importância biomédica
O conhecimento a respeito do metabolismo dos xenobióticos é fundamental para o
entendimento racional da farmacologia e terapêutica, toxicologia, pesquisa do cancro e vício
em fármacos. Todas estas áreas envolvem exposição a xenobióticos.
O metabolismo dos xenobióticos ocorre em 2 fases:
Na fase I, a principal reacção é a hidroxilação, catalizada pelos citocromos P450.
Na fase II, os compostos hidroxilados são convertidos por enzimas específicas em vários
metabolitos polares, através da conjugação com diversas substâncias. A fase II é responsável
pelo aumento da solubilidade (polaridade) e desta forma facilitar a excreção pelo organismo.
Isto sucede pois xenobióticos muito hidrofóbicos persistiriam no tecido adiposo quase que
indefinidamente, se não fossem convertidos em formas mais polares.
O termo “destoxificação” não é o mais apropriado, pois as reacções a que os xenobióticos
são sujeitos aumentam, por vezes, a sua toxicidade aumentando a sua actividade biológica.
55
3.4- Proteínas Celulares
A síntese proteica e um processo complexo que ocorre no citosol, mais precisamente
nos ribossomas. Estes são constituidos por proteínas e RNA ribossomal organizados em duas
subunidades, uma 60S e outra 40S. Nao vamos falar do processo de sintese em si mas dos
destinos das proteínas que daqui advêm.
As proteínas destinadas a secreção, integração nas membranas plasmaticas ou inclusão
em lisossomas partilham os primeiros passos de uma mesma via, que se inicia no retículo
endoplasmatico, explicada mais detalhadamente de seguida. As proteínas destinadas às
mitocôndrias, cloroplastos ou núcleo usam mecanismos separados que também irão ser
abordados. Para além destas existem outras proteínas que simplesmente se mantém no local
de síntese (citosol).
A sequência sinal é o elemento mais importante em muitas destas vias permitindo
“marcar” as proteínas consoante o seu destino. Para muitas proteínas esta é removida após a
chegada ao destino ou mesmo durante o transporte. Em proteínas com destino às mitocondrias
ou retículo endoplasmático esta sequência localiza-se no residuo N-terminal da sequência
polipeptídica.
Analisando especificamente a sequência sinal que encaminha proteínas para o RE
temos que a composição das sequências é mais ou menos sempre a mesma, tendo um ou mais
resíduos aminoacidos carregados positivamente proximos do N-terminal da sequência que
precedem a cadeia hidrofóbica de cerca de 10 a 15 resíduos. O C-terminal da sequência é
composto por resíduos polares, terminando no local de clivagem por onde a sequência vai ser
separada da cadeia.
O primeiro passo do direccionamento das proteínas para o RE começa com a síntese
proteica nos ribossomas, com o aparecimento da sequência sinal logo numa fase inicial, no
residuo N-terminal (por onde se inicia a síntese). Ao emergir do ribossoma, a sequência sinal
e o próprio ribossoma ligam-se à SRP (partícula de reconhecimento de sinal). A SRP junta-se
ao GTP ligando-se de seguida a receptores na membrana do RE. O peptido é ‘recebido’ pelo
complexo de translocação peptídica sendo no passo seguinte o SRP desligado do ribossoma
(dando-se a hidrólise do GTP). Continua o alongamento do peptido com o complexo a puxálo para dentro do RE (O ATP permite este processo) ate a proteína estar completamente
sintetizada. A sequência sinal é removida dentro do RE por uma sinal peptidase, dando-se a
dissociação e posterior “reciclagem” do ribossoma.
No lúmen do RE as proteínas continuam a sua modificação, havendo para além da
remoção de sequências sinais, alteração da conformação dos polipeptidos, formação de
ligações dissulfito entre as cadeias e glicosilação de proteínas que irá ser abordada mais à
frente.
De seguida, as proteínas são transportadas para o complexo de golgi em vesículas de
transporte onde, para além de haverem mais modificações a nível das glicoproteínas, vai
haver distinção e envio das proteínas para os respectivos destinos (por meios ainda não muito
bem compreendidos). A nível deste complexo, as proteínas que vão para as membranas,
secreções ou lisossomas têm de ser distinguidas por análise estrutural, já que já não existem as
sequências sinal. Este processo é melhor compreendido no caso das hidrolases destinadas aos
lisossomas, que são “marcadas” por fosforilação dos seus resíduos de manose.
Para direccionar as proteínas mitocondriais e dos cloroplastos também são necessárias
sequências sinal, mas este direccionamento só se inicia depois de uma proteína precursora ser
sintetizada e libertada do ribossoma. Esta proteína está ligada a proteínas chaperone do
citosol, sendo ‘entregue’ à membrana do respectivo organito. Depois disto, mecanismos de
translocação especializados transportam a proteéna ate ao seu destino sendo a sequência sinal
removida já no destino final.
Em relação ao núcleo, sabemos que as proteínas ribossomais são sintetizadas nos
ribossomas, vão para o núcleo para serem agrupadas em subunidades ribossomais,
regressando depois ao citosol. Por outro lado, os ribossomas sintetizam diversas proteínas
56
nucleares (RNA e DNA polimerases, histonas, topoisomerases etc) que têm de ser importadas
para o nucleo. Este tráfego é modulado por um sistema complexo de sinais moleculares e
proteínas de transporte.
Sabemos que o envelope nuclear rebenta em cada divisão celular e que após o
restabelecimento deste, as proteínas têm de ser reimportadas. Sendo assim, a sequência sinal
que permite que isto aconteça, a NLS (sequência de localização nuclear) constituida por 4 a 8
resíduos de aminoacidos não é removida quando as proteínas chegam ao seu destino e pode
estar localizada em qualquer sitio da proteina. A importação das proteínas ao núcleo é
mediada por muitas proteínas, como as importinas beta e alfa e uma GTPase (mais chamada
de Ran). Um dímero de importinas alfa e beta funciona como um receptor de proteínas
destinadas ao nucleo, ligando-se ao NLS das proteínas que estao no citosol. Este complexo é
translocado através do envelope nuclear por um mecanismo dependente da Ran GTPase. As
importinas alfa e beta dissociam.se, separando-se a proteína da importina alfa já dentro do
núcleo.
Existem proteínas como a LDL, as hormonas peptídicas, as proteínas destinadas a
degradação entre outras que se ligam directamente a receptores endocíticos situados em
invaginações da membrana. Forma-se uma vesícula (endossoma) com as proteínas e os
receptores, havendo posteriormente separação destes sendo o seu destino variável.
As glicoproteínas são proteínas que apresentam cadeias de oligossacáridos, às quais
estão ligadas covalentemente. Estas fazem parte de uma classe chamada glicoconjugados. As
cadeias de oligossacáridos ligadas as proteínas são pequenas mas com uma grande
diversidade estrutural. Outra característica das cadeias oligossacáridos que é amplamente
aceite é de que dependendo dos seus constituintes em açúcares e das suas sequências e
conformações este codificam informação biológica. Por exemplo, os resíduos de manose 6fosfato dirigem as enzimas lisossomais recém sintetizadas aos lisossomas.
Existem cerca de 200 monossacáridos na natureza, mas apenas 8 são usualmente
encontradas nas cadeias de oligossacáridos das glicoproteínas, e são elas: a galactose, a
glicose, a manose, o ácido N-acetilneuramínico, a fucose, o N-Acetilgalactosamina, o Nacetilglicosamina e a xilose. A maioria das reacções de glicosilação envolvidas na biossíntese
de glicoproteínas utiliza os açúcares nucleotídicos, e não os açúcares “sozinhos”. Isto porque
a natureza anidrido da ligação entre o grupamento fosfato e os açúcares é do tipo de altaenergia e de potencial elevado de transferência do grupo. Os açúcares deste compostos
encontram-se portanto “activados”, podendo ser transferidos para receptores específicos,
desde que existam transferases apropriadas.
Como muitas reacções de glicosidação ocorrem no lúmen do complexo de Golgi, é
necessário haver sistemas que transportem os açúcares nucleotídicos através da membrana.
Um desses sistemas é o antiporte, o afluxo de um açúcar nucleotídico é contrabalançado pelo
efluxo do nucleótido correspondente.
Existem três tipos principais de glicoproteínas, consoante a natureza das suas ligações, que
são: as O-ligadas, as N-ligadas e as que estão ligadas a um aminoácido carboxil terminal de
uma proteína, via um resíduo de fosforiletanolamina unido a um oligossacárido, que por sua
vez está unida a um fosfatidilinositol por uma glicosamina. Para além destas três classes
principais também existem outras proteínas que contém mais de um tipo de ligação.
As ligações O-glicosídicas são feitas entre o grupo hidroxilo da serina ou treonina e
um açúcar. Sendo que o açúcar a que se vai estabelecer a ligação mais predominante é o NAcetilgalactosamina. Os açúcares são doados pelos respectivos açúcares nucleotídicos,
reacções que são catalisadas pelas glicosiltransferases. A montagem das cadeias O-ligadas
envolvem estas enzimas que estão distribuidas sequencialmente na membrana do complexo
de Golgi, principalmente no compartimento do trans-Golgi. Um exemplo de glicoproteínas
com este tipo de ligações são as mucinas.
As ligações N-glicosídicas são feitas entre o nitrogénio amida da asparagina e a NAcetilglicosamina. A sua biossíntese envolve o processo intermédio de formação do
oligossacárido-pirofosforildolicol (Glc3Man9GlcNAc2-P-P-Dol), que posteriormente faz uma
57
ligação N-glicosídica com um ou mais resíduos de asparagina. Ocorrendo por fim o
processamento da cadeia de oligossacárido. Dependendo donde pára esse processamento, e
como ocorre, podem haver três classes de oligossacáridos: complexas, ricas em manose ou
híbridas.
No caso das cadeias ricas em manose, apenas os resíduos de glicose acrescidos de
determinados resíduos de manose são removido, sendo que o processamento da cadeia acaba
aí, apresentando normalmente 2 a 6 resíduos de manose. As cadeias complexas contém
geralmente resíduos terminais ácido N-acetilneuramínico e resíduos subjacentes de galactose
e N-acetilglicosamina, sendo primeiro removidos no retículo endoplasmático os resíduos de
glicose e os quatro de manose. Por fim, as cadeias híbridas contêm características das duas
classes.
O processo de N-glicosilação pode ser dividido em duas etapas. Na primeira etapa
existe a formação do oligossacárido-P-P-Dolicol através de um lípido chave que actua como
receptor de açúcares. Na segunda etapa existe o processamento da cadeia de oligossacárido
(que não será abordado neste trabalho).
O dolicol é um poliseprenol que é utilizado pelos tecidos eucariotas na biossíntese das
ligações anteriores. Este é após a borracha o hidrocarboneto mais longo de ocorrência natural
e de unidades simples. Antes de participar na biossíntese o dolicol é fosforilado a dolicolfosfato. Depois de fosforilado ele participa na reacção em que se forma o GlcNAcpirofosforil-dolicol (o lípido chave). Esta reacção ocorre na membrana do retículo
endoplasmático.
Depois da obtenção do lípido seguem-se quatro passos em que existe adição de
açúcares ao lípido, até se obter o oligossacárido-P-P-dolicol. No caso específico em que se
obtém Glc3Man9GlcNAc2-P-P-Dol, seguem-se os seguintes passos: no primeiro existe a
adição do segundo GlcNAc, usando o UDP-GlcNAc como doador; a seguir cinco resíduos de
manose são adicionados utilizando GDP-Manose; depois existe a adição de mais quatro
resíduos de manose mas desta vez o doador é o Dol-P-Man; por fim existe a adoção de três
resíduos de glicose periférica que são doados pelo Dol-P-Glc.
Depois de ser formado, o oligossacárido-P-P-Dolicol é transferido em bloco para a
superfície luminal da membrana do retículo endoplasmático, onde se vai ligar por uma ligação
N-glicosídica à asparagina. Esta reacção é catalisada pela oligossacárido: proteína transferase.
Um outro produto da reacção é o dolicol-P-P, que pode ser convertido em dolicol fosfato por
uma fosfatase, podendo ser utilizado novamente para o inicio da formação do oligossacárido.
A biossíntese descrita anteriormente é do tipo enzimática, sendo controlada por
diversos factores, entre os quais: o nível tecidual de dolicol-fosfato e a actividade da
oligossacárido transferase.
Existe ainda a biossíntese de ligações N-glicosídicas do tipo não enzimática –
glicação. Quando existem grandes concentrações de açúcares redutores, estes podem sofrer
uma reacção de adição nucleofílica com os grupo amino livres das proteínas formando iminas.
Este é o passo inicial da reacção de Maillard, onde existe então a formação da ligação Nglicosídica (fig. seguinte – o H2NR é a proteína):
É natural que as glicoproteínas, sendo agregados de cadeias oligossacáridas e de
polipéptidos, tenham a capacidade de desempenhar várias funções e ter várias acções de
âmbitos vários.
Para começar, a própria ligação covalente da proteína a um ou mais oligossacáridos induz
várias alterações na mesma:
58
•
•
A parte glicolítica modula propriedades físico-químicas da proteína, propriedades
essas que podem ser muito variadas (tamanho, estabilidade ao calor ou tempo de semivida em circulação)
Para além disso, as glicoproteínas tendem a proteger as proteínas a que estão
conjugadas da acção das proteases, evitando assim a sua proteólise.
No entanto, as funções das glicoproteínas não se restringem à acção que têm sobre a parte
proteica, e a cadeia oligossacárida por si, tem capacidades mais amplas. Esta funciona como
um código, um código de açúcares, que pode incluir informação sobre processos como
interacção entre células, desenvolvimento de tecidos e processamento de sinais extracelulares e, dentro da célula, tem papel fundamental no destino e processamento de
proteínas intracelulares no complexo de Golgi.
De uma forma mais geral, as glicoproteínas podem ainda desempenhar as seguintes
funções:
• Coagulação do sangue: tem um papel na resposta imunitária, os anticorpos são
glicoproteínas;
• Podem ter uma função estrutural, formando o colagénio, que é grande parte do
tecido conjuntivo no nosso corpo;
• Função hormonal: há hormonas que são glicoproteínas, como e o caso da hormona
estimulante da tiróide;
• Função enzimática: um exemplo e a fosfatase alcalina, existente em vários órgãos;
• As glicoproteínas podem ser parte integrante de membranas celulares: as cadeias
de carbo-hidratos podem ligar-se às proteínas integrantes da membrana e formar
glicoproteínas que depois têm um papel fundamental nas interacções entre células ou
entre a célula e a matriz extra-celular.
• Podem funcionar como agentes lubrificantes e protectores, no caso das mucinas. Estas
vão ser responsáveis pelo bloqueio da interacção entre bactérias ou qualquer outro tipo
de “ataque” com a superfície da célula a que estão ligadas, num mecanismo que
acontece, por exemplo, no estômago. A produção de muco impede a auto-digestão por
parte das várias enzimas presentes e o ataque do meio ácido.
As glicoproteínas têm ainda a capacidade de interagir com lectinas e selectinas, que lhes
confere funções bastante mais específicas e abrangentes. A parte de carbo-hidratos que se liga
a uma proteína contem informação importante que intervém em variados processos, como já
foi referido. Essa informação está altamente organizada, é muito complexa e como tal,
necessita de algo que a saiba “ler” e interpretar, à semelhança do que acontece com o código
genético. Vão ser as lectinas que vão ser responsáveis por essa interpretação e vão-se ligar à
cadeia oligossacárida com grande afinidade e especificidade. Depois da interpretação deste
código, vão ser capazes de mediar ou desencadear uma grande variedade de processos e
funções intra e extra-celulares. Como exemplos de aplicação das lectinas, e só a título de
exemplo, temos que estas intervêm no mecanismo de renovação dos eritrócitos (nos
mamíferos), na absorção de galactose nos hepatócitos e na interacção de vírus e bactérias com
as células hospedeiras.
Dentro da família destas lectinas, existem as selectinas.
Situadas na membrana plasmática, as selectinas mediam o
reconhecimento entre células e adesão celular no âmbito de
vários processos, como o movimento de células
imunitárias, os linfócitos T, do sangue para o sítio
específico que se encontra infectado ou inflamado.
Assim, um linfócito T livre, circulando num capilar,
sofre interacções nos ligandos de glicoproteínas da sua
superfície, acção das selectinas que se encontram no
endotélio dos capilares. Estas interacções vão atrasar o
movimento do linfócito e fazem-no rolar pela superfície
59
externa do capilar; quanto mais perto o linfócito está do local-alvo, mais fortes vão ser as
interacções até ao momento em que o ligando na superfície do linfócito T promove a adesão
celular. Nesse momento, a célula T pára, e sobre a influência de sinais vindos do próprio local
de inflamação, começa o processo de extravasão: o linfócito entra através da parede do
capilar.
As lectinas também agem em processos dentro da célula, um destes exemplos foi a já
falada distribuição de proteínas recentemente sintetizadas no complexo de Golgi. Um resíduo
de manose de um oligossacárido de uma glicoproteína é fosforilado através de uma enzima
que o reconhece, e, preferencialmente no fim da cadeia de carbo-hidratos, catalisa a reacção
de manose a manose-6-fosfato. O resíduo fosforilado vai ser depois reconhecido através de
uma lectina no lado luminal do complexo de Golgi. Esta “etiquetação” das proteínas vai
permitir depois a sua transferência para os lisossomas.
Este processo envolve o processamento da grande maioria das enzimas de degradação, as
hidrolases.
60
3.5- Proteínas Plasmáticas
Origem, Tipos Principais e Funções
O plasma sanguíneo é constituído aproximadamente por 90% de água e 10% de solutos, de
entre os quais 70% correspondem a proteínas plasmáticas; estas podem ser proteínas simples,
glicoproteínas (quase na totalidade) ou lipoproteínas.
Os tipos principais de proteínas plasmáticas são: albumina, globulinas, lipoproteínas e
fibrinogénio.
A grande maioria das proteínas plasmáticas é sintetizada no fígado, sendo as restantes
produzidas em células plasmáticas (γ-globulinas) ou noutros tecidos, como as células
endoteliais. A síntese destas proteínas ocorre em polirribossomas ligados à membrana do
retículo endoplasmático rugoso, ficando ligadas ao sistema membranar. As proteínas seguem
então para o retículo endoplasmático liso, depois para o complexo de Golgi, no qual são
incorporadas em vesículas secretoras, e finalmente eliminadas para o plasma. Geralmente, as
proteínas são sintetizadas na sua forma zimogénio, sofrendo depois modificações à medida
que são transportadas para o exterior da célula, para adquirirem actividade biológica
específica. As proteínas plasmáticas apresentam, frequentemente, polimorfismo.
A albumina é a proteína mais abundante no plasma sanguíneo. É sintetizada na sua forma
zimogénio – prealbumina, sofrendo clivagem peptídica à medida que é transportada para o
plasma. A sua concentração é um factor determinante na manutenção da distribuição de
fluídos entre o sangue e o espaço intersticial tecidular, através do equilíbrio entre as pressões
hidrostática vascular e osmótica. O facto de ser carregada negativamente é de extrema
importância, pois evita que seja filtrada a nível dos glomérulos de Malpighi, cuja membrana
basal é também carregada negativamente. Possui uma parte apolar, a qual permite a ligação de
moléculas como ácidos gordos de cadeia longa, bilirrubina, fármacos, hormonas esteróides,
vitaminas, e ainda iões como Ca2+ e Mg2+, actuando como transportador destes compostos
pelo sangue.
As globulinas abrangem uma vasta gama de proteínas globulares. Podem ser divididas em
quatro classes: α1, α2, β e γ. As globulinas α1, α2 e β estão envolvidas no transporte de lípidos,
hormonas, vitaminas e iões metálicos. O grupo γ consiste nas imunoglobulinas, moléculas
centrais dos mecanismos de acção do sistema imunitário (incluem os vários tipos de
anticorpos e os receptores de membrana dos linfócitos).
61
Classe α1:
Proteínas
Antitripsina
Antiquimotripsina
HDL
Protrombina
Transcortina
Função
Inibição da tripsina e outras proteases
Inibição da quimotripsina
Transporte de lípidos
Precursor da trombina
Transporte de cortisol, progesterona e
corticosterona
Transporte de progesterona
Transporte de iodotironinas
Glicoproteína ácida
Globulina de ligação da tiroxina
Classe α2:
Proteínas
Ceruloplasmina
Antitrombina III
Haptoglobina
Função
Transporte de iões cobre
Inibição da coagulação
Ligação da hemoglobina, diminuindo
a sua actividade oxidativa para
posterior degradação no sistema
reticuloendotelial
Cisão de ésteres de colina
Precursor da plasmina – fundamental
na
degradação
de
proteínas
plasmáticas
Ligação de proteases e transporte de
iões zinco
Transporte de vitamina A
Colinesterase
Plasminogénio
Macroglobulina
Proteína de ligação do retinol
Classe β:
Proteínas
Função
LDL
Transporte de lípidos
Transferrina
Transporte de iões de ferro
Globulina de ligação de hormonas Transporte de testosterona e estradiol
sexuais
Transcobalamina
Transporte de vitamina B12
Proteína C-reactiva
Activação do sistema complemento
Classe γ:
Proteínas
IgG
IgA
IgM
IgD
IgE
Função
Imunidade mediada por anticorpos
Prevenção da infecção de mucosas
Imunidade mediada por células B
Receptores membrares das células B
Resposta alérgica
62
As lipoproteínas presentes no sangue são o meio de transporte para os lípidos que não são
transportados dissolvidos no plasma nem ligados à albumina. As lipoproteínas são agregados
esféricos ou discóides de lípidos e apoproteínas. Consistem num núcleo de moléculas apolares
envolto numa monocamada lipídica de moléculas anfipáticas. A sua classificação baseia-se na
sua densidade: quanto maior a quantidade de lípidos apolares no seu núcleo, menor a sua
densidade. Existem 5 tipos de lipoproteínas (por densidade crescente): quilomicra, VLDL,
IDL, LDL, HDL. A quantidade de apoproteínas aumenta com a densidade (maior nas HDL), e
têm a função de marcadores superficiais.
Os quilomicra transportam sobretudo triacilgliceróis (daí a sua baixa densidade) do
intestino delgado para os tecidos; as VLDL, IDL e LDL (são formadas como VLDL,
adquirindo gradualmente densidade transformando-se em IDL e depois LDL) são
especializadas no transporte de colesterol, fosfolípidos e triacilgliceróis (em menor
quantidade que os quilomicra) de uns tecidos para outros; por último as HDL são
responsáveis pela mobilização do excesso de colesterol dos tecidos para o fígado, onde é
armazenado.
O fibrinogénio é uma glicoproteína sintetizada nos hepatócitos e nos megacariócitos
(células da medula óssea responsáveis pela produção de plaquetas sanguíneas); é solúvel em
água, e consiste na forma precursora da fibrina (insolúvel), responsável pela formação de
coágulos sanguíneos. A proteína trombina é responsável pela conversão do fibrinogénio em
fibrina, em situação de ruptura do vaso sanguíneo.
Metabolismo do Ferro
O ferro é um mineral indispensável ao organismo, visto ser o constituínte central dos
grupos heme, permitindo o transporte de oxigénio, e de citocromos, que intervêm em reacções
de oxidação-redução. É ainda constituínte de muitas enzimas, ou actua como cofactor destas.
O metabolismo do ferro é mais importante nas mulheres do que nos homens, devido à
menstruação, lactação e gestação. Cerca de 20 mL de eritrócitos são catabolizados por dia, o
que resulta em 25mg de ferro. Porém, o sistema reticuloendotelial é capaz de reciclar a sua
maioria para formar novos glóbulos vermelhos.
Não existe mecanismo fisiológico para a excreção do ferro, dái a extrema importância da
regulação da absorção para controlo das quantidades de ferro no organismo. A única excreção
feita (e mesmo assim, quase insignificante) é através da sudação, escamação de pele e
menstruação.
Transferrina
A transferrina é uma glicoproteína do plasma sintetizada pelo fígado,que tem como
função o transporte do ferro para os locais onde este é necessário. Quando não está ligado a
esta molécula, o ferro, por ser tanto receptor como dador de electrões (muito reactivo), tem
tendência a transformar o peróxido de hidrogénio em radicais livres (elementos altamente
prejudiciais à vida celular).
A esta molécula podem ligar-se dois iões férricos (Fe3+).
A sua concentração no plasma é de 300mg/dL. Normalmente, encontra-se a um terço da
saturação máxima. A capacidade total de ligação do ferro do plasma é de 300 µg/dL.
Existem, na superfície de muitas células, receptores (TfR) sensíveis à transferrina. Quando
existem formação do complexo transferrina-TfR, a transferrina passa para dentro da célula por
endocitose mediada por receptor. O pH ácido existente no lisossoma, irá promover a
dissociação do ferro. Este vai para o citoplasma através de um Transportador de Metais
Divalentes, o DMT1, e a apotransferrina retorna à membrana plasmática, dissocia-se do
receptor, reentra no plasma e liga-se a outro ião de ferro.
63
Absorção do ferro a nível do intestino delgado
Diariamente, o Homem ingere cerca de 10 a 20 mg de ferro. Porém, somente 1 a 2mg é
absorvido no intestino, mais concretamente, no duodeno proximal e, em menor quantidade, no
jejuno superior. A absorção ao nível dos enterócitos pode ser na forma de iões ou grupos
heme.
Na superfície dos enterócitos, existe uma enzima, a ferriredutase (por vezes a vitamina C
também pode adquirir esta função), que catalisa a reacção:
Fe 3 Fe 2
Com a ajuda de um
Transportador de Metais Divalentes
(DMT1), o ião ferro passa para dentro do eritrócito. Aí, ou se liga à ferritina ou é transferido
através da membrana basolateral do enterócito para o plasma. Esta passagem é permitida por
uma ferroportina, uma proteína reguladora (IREG1), que interage com a Hefastina (esta
contém cobre e permite a reacção inversa da ferriredutase). O ião férrico vai posteriormente
ser transportado pela tranferrina.
Quando o ferro extracelular se encontra associado a um grupo heme, este grupo passa para
dentro da célula através de uma Transportadora de Heme e, dentro do enterócito, a enzima
heme oxidase remove-lhe o ião ferroso.
Quando já foi ingerido suficiente ferro, o enterócito bloqueia a absorção do mesmo
(mecanismo de controlo da absorção). Este acontecimento também pode surgir em resposta à
necessidade ou não de ferro para realizar a eritropoiese.
Ferritina
Uma molécula de ferritina é capaz de armazenar entre 4000 e 4500 iões de ferro. E
encontra-se no citoplasma Em média, armazena cerca de 23% do ferro corporal (a grande
maioria encontra-se ligada a grupos heme). Excepto em situações patológicas, existem em
pequena quantidade no plasma.
A Hemossiderina - ferritina parcialmente degradada ainda ligada a iões de ferro também
pode estar presente; a sua concentração aumenta nos tecidos em situações patológicas.
Patologias associadas
Anemia por défice de ferro
Pode ser devida a: hábitos alimentares pobres, perda, má absorção ou utilização indevida
de ferro.
Hemocromatose
Doença autossómica recessiva
Existe um excesso de ferro nos tecidos (acima de 15g quando o normal é entre
2,5 e 3,5)
A concentração de ferritina é muito elevada, em especial no fígado e baço.
O ferro em excesso começa a atacar os organelos celulares, nomeadamente as
mitocôndrias, produzindo danos que podem levar a dano e mesmo morte celular.
A quantidade de hemosiderina aumenta nos tecidos.
Hemocromatose secundária: pode ocorrer após transfusões sanguíneas ou
consumo excessivo de ferro.
64
Imunoglobulinas
São glicoproteínas que conferem imunidade humoral ao ser humano. Também chamadas
de Anticorpos, representam 15-20% das proteínas plasmáticas. As Imunoglobulinas são
síntetizadas e excretadas por células plasmáticas, derivadas de linfocitos B, no sistema
retículoendotelial.
Estrutura:
São moléculas simétricas, formadas por 4 cadeias polipeptídicas: 2 leves (L) e 2
pesadas (H).
Tem uma região constante, e uma região variável (sequência variável de aminoácidos)
que é o local de ligação do antigénio.
As 4 pontes dissulfureto ajudam a manter a estrutura quaternária, enquanto que as
outras ligações, intracadeias (de Cl2 a Cl4 e de Cl1 a VL), mantêm a estrutura terciária
das cadeias. Formando uma proteína globular em forma de Y.
Na parte constante, a região Cl2 delimitada por duas pontes dissulfureto é chamada de
Região Hinge, que permite a mobilidade dos braços, sendo sensível à proteólise pela
pepsina e papaína;
Há cinco classes de imunoglobulinas com função de anticorpo:
IgM – é a primeira imunoglobulina a ser expressa na membrana do linfócito B durante
o seu desenvolvimento. Na membrana das células B, a IgM está na forma monomérica e
no plasma sanguíneo apresenta-se como pentâmero. É o primeiro anticorpo a ser
produzido numa resposta imunitária primária.
IgA – é a imunoglobulina predominante nas secreções mucosserosas (saliva, nasais,
lágrimas, leite, etc.). O principal papel da IgA é proteger o organismo da invasão viral
ou bacteriana. Mais de 80% aparece sobre a forma monomérica no sangue e apresenta
forma dimérica nas restantes secreções.
IgG – é a imunoglobulina mais abundante no plasma (70%-75%) e está distribuída
uniformemente entre os espaços intra e extravasculares (possui mobilidade através das
65
paredes dos vasos capilares). Sendo a mais importante da resposta imunitária
secundária, é a unica com capacidade de atravessar a barreira placentária, conferindo
um alto grau de imunidade passiva ao feto e recém-nascido.
IgE – é encontrada nas membranas superficiais dos mastócitos e basófilos em todos os
indivíduos. Esta classe de imunoglobulinas sensibiliza as células nas superfícies das
mucosas conjuntiva, nasal e brônquica. Responsável pelas manifestações fisiológicas da
alergia, nesta situação aumenta a sua frequência no plasma.
IgD – tendo função desconhecida encontra-se em menos de 1% no plasma. Presente na
superfície de quase todas as células B maduras.
66
3.6- Hemostase, hemoglobina e transporte de gases
pelo sangue
Proteínas da Hemostase
O termo hemostase está relacionado com a ‘estase’ do sangue, isto é, com a
manutenção do mesmo em circulação, tanto pela prevenção da extravasão – perda – de
sangue, como pelo impedimento da formação de trombos/coágulos indesejados. De facto,
sempre que ocorre a ruptura de um vaso, com o objectivo de atingir a hemostase, recorre-se a
quatro mecanismos: constrição vascular, formação de um rolhão de plaquetas, formação de
um coágulo sanguíneo e, por último, o crescimento de tecido fibroso (que pode não ser
necessário).
O processo de coagulação consiste numa série de reacções químicas que culminam na
formação da dita rede de fibrina. Para além das plaquetas, os factores de coagulação mais
importantes podem designar-se através da numeração romana, como factores I a XIII (não
existe o VI), que se irão activar mutuamente (passando a ter um ‘a’ agregado ao nome neste
caso), numa cascata de reacções (ver tabela 1). Note-se que o número associado a cada factor
não está relacionado com a ordem de actuação dos mesmos, mas sim apenas pela ordem por
que foram descobertos.
A coagulação dá-se em três fases
(ver esquema):
1.
Formação do Activador
da Protrombina – pode dar-se
pela via extrínseca ou pela via
intrínseca;
2.
A
protrombina
é
activada em trombina;
3.
A trombina, por sua vez,
converte o fibrinogénio solúvel
(outra proteína plasmática
formada no fígado) em fibrina
insolúvel, que formará a parte
mais exterior do coágulo.
Tabela 1 - Sistema numérico de
nomenclatura
dos
factores
coagulação
Factor Nome Comum
I
Fibrinogénio
II
Protrombina
III
Factor Tissular (TF)
IV
Ca2+
V
Pro-acelerina
VII
Pro-convertina
VIII
Globulina anti-hemofílica
(AHG)
de
67
IX
Componente
Tromboplastina do Plasma
(PTC)
X
Factor Stuart-Prower
XI
Antecedente da
Tromboplastina do Plasma
(PTA)
XII
Factor Hageman
XIII
Factor Estabilizador da
Fibrina (FSF)
Há duas vias iniciais para a formação do coágulo sanguíneo. Na via intrínseca, mais
lenta e complexa, é o contacto do sangue com as fibras de colagénio afectadas, em conjunto
com as plaquetas activadas, que activa os factores de coagulação. A via começa com a ‘fase
de contacto’, na qual a precalicreína (PK), o cininogénio (HK) e os factores XII e XI são
expostos a uma superfície activante carregada negativamente. O factor XII é activado em XIIa
por protólise pela calicreína, e, por sua vez, induz a transformação de precalicreína em
calicreína, num processo de activação recíproca. Por outro lado, o factor XIIa activa o factor
XI, com a libertação de bradicinina (um nonapéptido de forte poder vasodilatador), a partir do
cininogénio. Na presença de Ca2+, o factor XIa activa o factor IX numa serina-protease que,
por sua vez, cliva uma ligação Arg-Ile no factor X, activando-o numa serina-protease de dupla
cadeia, Xa. Para esta última reacção, é formado o complexo tenase na superfície das plaquetas
activadas, que têm na sua superfície externa os fosfolípidos (PL) carregados negativamente,
fosfatidilserina e fosfatidilinositol (normalmente na superfície plasmática interna). Note-se
que, em todas as reacções envolvendo zimogénios que contenham γ-carboxiglutamato, os
terminais amina servem como locais de ligação com muita afinidade para o Ca2+. O factor
VIII, uma glicoproteína, é um cofactor que funciona como um receptor na superfície das
plaquetas dos factores IXa e X. Este é activado pela trombina em VIIIa que, por sua vez, é
inactivado pela degradação da trombina.
A via extrínseca, mais rápida e directa, tem este nome porque são as células
subendoteliais afectadas pela ruptura que libertam a proteína TF (factor III), para a corrente
sanguínea. Com a ajuda de factores de coagulação e de Ca2+ (factor IV), essenciais para todo o
processo, a TF é convertida em protombinase. A via começa então com a interacção entre o
TF e o factor VII, produzido no fígado, que é activado no processo em VIIa. O TF actua como
um cofactor para o factor VIIa (formando o complexo factor tissular), melhorando a
actividade enzimática deste, de activação do factor X, por clivagem da ligação Arg-Ile
(análoga à acção do complexo tenase na via intrínseca). A activação do factor X é um elo de
ligação importante entre as duas vias de formação do activador da protrombina. Para além
deste, temos que a complexação do factor tissular com o factor VIIa vai também activar o
factor IX na via intrínseca, sendo, por isso, considerado o passo-chave da coagulação em seres
vivos.
O TFPI (tissue factor pathway inhibitor) é um inibidor da coagulação muito importante.
Trata-se de uma proteína que circula no sangue associada a lipoproteínas e que inibe
directamente o factor Xa por ligação à enzima no local activo. O complexo factor Xa-TFPI,
por sua vez, inibe o complexo factor VIIa-TF.
Os eventos que se dão abaixo do factor Xa são designados via final comum. Tanto as
vias extrínseca e intrínseca, como a via final comum envolvem uma série de proteínas
diferentes, que podem ser classificadas em cinco tipos (ver tabela 2): (1) zimogénios de
proteases serina-dependentes, que se tornam activas durante o processo; (2) cofactores;
68
(3) fibrinogénio; (4) transglutaminase, que estabiliza a rede de fibrinas; e (5) proteínas
reguladoras e outras.
Tabela 2 – Funções das proteínas envolvidas na coagulação do sangue
Zimogénios de Serina Proteases
Factor XII
Liga-se à parede endotelial da superfície afectada, carregada negativamente;
activado pelo cininogénio e calicreína (elevados pesos moleculares)
Factor XI
Activado pelo factor XIIa
Factor IX
Activado pelo factor XIa na presença de Ca2+
Factor VII
Trombina activada na presença de Ca2+
Factor X
Activado na superfície das plaquetas activadas pelo complexo tenase (Ca2+,
factores VIIIa e IXa) e pelo factor VIIa na presença de TF e Ca2+
Factor II
Activado na superfície das plaquetas activadas pelo activador da protrombina
Nota: os factores II, VII, IX e X são zimogénios que contêm γ-carboxiglutamato
Cofactores
Factor VIII
Activado pela trombina; o factor VIIIa é o cofactor na activação do factor X
pelo factor IXa
Factor V
Activado pela trombina; o factor Va é o cofactor na activação da
protrombina pelo factor Xa
TF (Factor
III)
Uma glicoproteína existente na superfície endotelial das células afectadas,
que funciona como cofactor para o factor VIIa
Fibrinogénio
Factor I
Clivado pela trombina para formar o coágulo sanguíneo
Transglutaminase Dependente do Tiol
Factor XIII
Activado pela trombina na presença de Ca2+; estabiliza as fibras de
fibrina por criação de ligações cruzadas
Proteínas Reguladoras e Outras
Proteína C
Activada em proteína Ca pela ligação da trombina à trombomodulina;
degrada os factores VIIIa e Va
Proteína S
Actua como cofactor da proteína C; ambas possuem resíduos de
γ-carboxiglutamato
Trombomodulina
Encontra-se na superfície das células endoteliais; agrega-se à trombina,
que por sua vez activa a proteína C
O activador da protrombina, também designado complexo protrombinase, é
constituído por fosfolípidos carregados negativamente, protrombina, cálcio e factores Va e
Xa. O activador da protrombina tem a função que o seu nome indica, convertendo a
protrombina em trombina (que tem uma actividade protolítica importantíssima), na superfície
das plaquetas activadas. É o contacto das plaquetas com as fibras de colagénio que
69
desencadeia uma série de processos bioquímicos que activam a scramblase, responsável pelo
transporte dos ditos fosfolípidos, que vão permitir a formação do activador da protrombina.
A protrombina (factor II) é uma proteína plasmática, formada continuamente no fígado
com o auxílio da vitamina K. Como se trata de uma proteína instável, divide-se facilmente em
compostos mais pequenos, como é o caso da trombina.
O fibrinogénio (factor I) é uma glicoproteína plasmática constituída por três cadeias
polipeptídicas diferentes (Aα,Bβγ)2, todas sintetizadas no fígado, ligadas covalentemente por
pontes dissulfito. Os três genes estruturais responsáveis encontram-se no mesmo cromossoma,
sendo a sua acção regulada coordenadamente nos humanos. As porções A e B das cadeias Aα
e Bβ (respectivamente fibrinopéptidos A, FPA, e fibrinopéptidos B, FPB) possuem no
terminal amina carga negativa, que se deve à presença de resíduos de aspartato e glutamato,
assim como de tirosina O-sulfato na FPB. É esta carga negativa que vai contribuir para a
solubilidade do fibrinogénio no plasma, assim como para prevenir a agregação de várias
moléculas de fibrinogénio, através de repulsão electroestática.
A trombina formada actua então sobre o fibrinogénio (factor I), provocando a hidrólise,
por molécula, de quatro ligações Arg-Gly entre os fibrinopéptidos e as porções α e β (das
cadeias Aα e Bβ) do fibrinogénio, originando monómeros de fibrina. A remoção dos
fibrinopéptidos torna expostos locais de ligação, o que induz a polimerização de vários destes
monómeros, formando-se fibras de fibrina longas.
A fibrina é uma proteína bastante flexível, de cor esbranquiçada, insolúvel, constituinte
essencial do coágulo sanguíneo. Nos estados iniciais da polimerização, os monómeros apenas
se encontram ligados por ligações covalentes de hidrogénio, pouco fortes. É então necessária
a acção do factor estabilizador da fibrina (factor XIII), uma transglutaminase, presente em
pequena quantidade nas globulinas plasmáticas e também libertado pelas plaquetas. A
trombina tem como função adicional a activação deste factor, que, então, favorece a formação
de cada vez mais ligações covalentes, assim como de ligações cruzadas entre os vários
monómeros, tornando a rede de fibrina numa estrutura tridimensional muito mais forte e
compactada, o que provoca uma retenção por aglutinação dos elementos figurados do sangue
(eritrócitos, plaquetas, leucócitos granulares e agranulares).
A concentração da trombina formada no processo de hemostase tem de ser
cuidadosamente controlada, por duas formas, de forma a evitar excessiva formação de fibrina
e activação de plaquetas. Por um lado, a trombina circula no sangue sob a sua forma
precursora (inactiva), protrombina, sendo que a cascata de reacções que precede a activação
deste zimogénio possui um delicado equilíbrio activação/inibição. Por outro lado, temos a
inactivação da trombina em si, que é conseguida através de inibidores presentes na circulação,
sendo o mais importante a protrombina III, uma α-globulina (responsável por cerca de 75%
do controlo). Para além deste, actuam também a α2-macroglobulina, a heparina cofactor II e a
α1-antitripsina.
Durante a formação de um coágulo, 85-90% da trombina formada é adsorvida pelas
fibras de fibrina ou combinada com a antitrombina III, prevenindo a extensão do coágulo em
demasia, pelo bloqueamento do efeito da trombina sobre o fibrinogénio. A antitrombina III
inibe os efeitos dos factores IXa, Xa, XIa, XIIa e VIIa (complexado com o TF) na via
intrínseca da coagulação. A acção desta proteína pode ter uma eficácia ainda muito mais
elevada pela sua combinação com a heparina, um anticoagulante importantíssimo, que se vai
ligar à trombina III num local catiónico, originando uma alteração conformacional que
promove a ligação desta à trombina e a outras substâncias. Assim, a trombina pára de
estimular a fragmentação do fibrinogénio, deixando de se formar os monómeros de fibrina e,
em último caso, o coágulo.
Existem ainda outros factores que contribuem para o sistema anticoagulante, tais como:
1.
O facto da parede endotelial ter uma superfície lisa, que previne a activação das
plaquetas;
2.
Os glicocálices do endotélio, superfície externa da membrana plasmática, que
repelem as plaquetas e os factores coagulantes;
70
3.
A trombomodulina, uma proteína ligada à membrana endotelial, que adere à
trombina, tornando o processo de coagulação mais lento e activando a proteína C.
Esta, combinando-se com a proteína S, forma a APC (activated protein C), degrada os
factores Va e VIIIa, limitando a sua acção coagulante.
Dada então a acção protectora dos anticoagulantes, tanto a fibrina não utilizada como a
pertencente ao coágulo (uma vez cumprida a sua função), são lisadas, através do mecanismo
fibrinolítico. Sendo as enzimas activadoras deste mecanismo as serina-proteases, os
inibidores do mesmo são, naturalmente, os serpin (serine-proteinase inhibitor), que se
acoplam à enzima alvo.
O principal agente da acção fibrinolítica é a plasmina, serina-protease de dupla cadeia,
enzima principalmente responsável pela degradação da fibrina e do fibrinogénio. Esta tem a
sua origem no plasminogénio, onde actuam activadores de dois tipos: alteplase (t-PA) e
urocinase (u-PA). A função comum destes é clivar a ligação Arg-Val, produzindo a plasmina
A alteplase, assim como o seu inibidor, PAI, são sintetizados e secretados pelas células
endoteliais, cujas superfícies têm afinidade tanto para o t-PA como para o plasminogénio, que
é activado a plasmina, fixada na superfície da fibrina (fase sólida). Note-se que a activação do
plasminogénio é cerca de cem vezes superior na fase sólida, relativamente à líquida (sangue
circundante). A alteplase é libertada na circulação em casos de formação de feridas ou sob
stress, não exercendo qualquer actividade se não se encontrar ligada à fibrina. Assim, a
alteplase está relacionada com a fibrinólise, processo responsável pela dissolução de
coágulos.
A urocinase, de origem urinária, é responsável por fenómenos de migração celular e
remodelação tecidular (degradação de matriz extracelular). A prourocinase é o precursor da
urocinase, que é sintetizada por monócitos, macrófagos, fibroblastos e células epiteliais.
Transporte de Oxigénio pelo Sangue
A circulação sanguínea é responsável pelo fornecimento de oxigénio a todos os tecidos
do corpo. No pulmão o oxigénio difunde-se dos alvéolos para o sangue dos capilares. Este
depois é bombeado do coração para os tecidos. O oxigénio é maioritariamente (97%)
transportado em associação com a hemoglobina presente nos eritrócitos segundo um
mecanismo já estudado anteriormente. O restante é transportado dissolvido no plasma
sanguíneo. É apenas através desta associação que se consegue fornecer todo o oxigénio
necessário ao bom funcionamento dos tecidos.
A difusão deste gás ocorre quando há diferenças de pressão parcial de oxigénio (Po 2)
entre duas zonas. Nos alvéolos Po2 é de 104 mm Hg enquanto nos capilares que chegam com
sangue venoso é de 40 mm Hg. O sangue que chega aos tecidos tem uma Po2 de 95 mm Hg
enquanto
os
tecidos
apresentam
uma
Po2
variável,
em
média
de
23 mm Hg. O oxigénio liberta-se da hemoglobina e difunde-se para os tecidos, saindo o
sangue venoso com uma Po2 de 40 mm Hg, igual à Po2 do interstício.
Por 100 ml de sangue temos 15g de hemoglobina e cada grama de hemoglobina
consegue-se ligar a 1.34 ml de oxigénio. Assim, temos que 100 ml de sangue conseguem
transportar cerca de 20 ml de oxigénio, se a hemoglobina estiver 100% saturada.
A capacidade de associação entre o
oxigénio e a hemoglobina pode ser quantificada.
O
oxigénio liga-se reversivelmente ao grupo heme
da
hemoglobina. Esta ligação é favorecida quando
Po2
é alta e quebra-se quando Po2 é baixa. A curva de
dissociação oxi-hemoglobina mostra o grau de
saturação da hemoglobina pelo oxigénio. A uma
Po2
de 95 mm Hg (ao sair do pulmão) a hemoglobina
está
97% saturada com oxigénio enquanto aos 40 mm
Hg
(sangue venoso) tem um grau de saturação de
apenas 75%. Podemos ainda considerar o valor
P50
71
como uma nova medida de afinidade relativa entre o oxigénio e a hemoglobina. O P50 é o
valor de Po2 para o qual metade da hemoglobina está saturada de oxigénio. A curva de
dissociação tem uma forma sigmóide. Isto deve-se à estrutura tetramérica da hemoglobina,
que permite que ela transite entre uma conformação de estado T (de pouca afinidade) para
uma de estado R (de grande afinidade) à medida que o oxigénio se vai ligando.
A curva de dissociação oxi-hemoglobina é afectada por diversos factores,
nomeadamente pH, [CO2], temperatura e [BPG] (2,3-bifosfoglicerato). A concentração de
protões, de dióxido de carbono e de BPG afectam na medida em que estes são efectores que
promovem a libertação de oxigénio, favorecendo a forma desoxigenada da hemoglobina.
Apesar de não ocuparem o sítio de ligação do oxigénio à hemoglobina, a sua ligação implica
mudanças conformacionais na hemoglobina que vão afectar a capacidade de ligação do
oxigénio. Uma vez que estes efectores alostéricos se ligam a sítios específicos da
hemoglobina os seus efeitos são cumulativos.
A concentração de CO2 e o valor de pH na hemoglobina estão directamente
relacionados, como veremos mais à frente. Quando o sangue fica ligeiramente mais ácido,
diminuindo do valor normal de pH de 7.4 para 7.2 verifica-se um desvio da curva de cerca de
15% para a direita. Um aumento de pH desvia a curva para a esquerda. Perto dos tecidos (por
estarem metabolicamente activos) a concentração de dióxido de carbono e de protões é
elevada. Quando o sangue chega aos capilares é favorecida a ligação destes efectores e, como
tal, a afinidade da hemoglobina para o oxigénio é menor (P50 diminui), favorecendo a sua
libertação. O resultado geral é que o oxigénio tem maior facilidade em se libertar da
hemoglobina para o plasma sanguíneo e daí para a mioglobina presente nos tecidos. Ao
regressar aos pulmões a concentração de protões e a pressão parcial de Co2 é baixa, o que
favorece a sua libertação da hemoglobina, aumentando a afinidade desta para o oxigénio. Este
efeito é conhecido como o efeito de Bohr, e pode ser explicado pela seguinte reacção:
HHb+ + O2
HbO2 + H+ , onde HHb+ é uma forma protonizada da
hemoglobina.
Um dos grandes contributos para o efeito de Bohr é provocado pelo His 146 da subunidade . Este resíduo, quando ligado ao H+ ajuda a estabilizar a hemoglobina no estado T,
diminuindo portanto a sua afinidade para o oxigénio e, consequentemente, o valor de P 50.
Assim, podemos dizer que à medida que a concentração de protões ligados à hemoglobina
aumenta verifica-se uma transição para o estado T, que favorece a libertação de oxigénio. A
protonização de outros resíduos tem um efeito semelhante.
O BPG é conhecido por diminuir a afinidade da hemoglobina para o oxigénio,
desviando a curva de dissociação oxi-hemoglobina para a direita. Tal como o dióxido de
carbono e o H+, a ligação do oxigénio e do BPG à hemoglobina está inversamente
relacionada, e pode ser explicado pela seguinte reacção:
HbBPG + O2
HbO2 + BPG
O BPG é uma pequena molécula sintetizada nos eritrócitos, que se liga a uma pequena
cavidade da hemoglobina desoxigenada (estado T). Na hemoglobina oxigenada (estado R)
esta cavidade é demasiado pequena para que a BPG se ligue. Quando a BPG está ligada à
hemoglobina desoxigenada estabiliza a sua conformação T, diminuindo a sua capacidade de
se ligar ao oxigénio e facilitando a libertação deste para os tecidos. Em situações de hipoxia
prolongadas (por exemplo a altitudes elevadas, onde Po2 é baixa) a concentração de BPG
aumenta. Este ajuste tem um efeito relativamente pequeno na ligação do oxigénio à
hemoglobina nos pulmões, mas nos tecidos é libertado muito mais oxigénio. Por isso é que
atletas que treinem nestas condições conseguem aumentar temporariamente a sua capacidade
aeróbica.
Um aumento da temperatura corporal também se traduz num desvio da curva de
dissociação para a direita, diminuindo a afinidade da hemoglobina para o oxigénio e
favorecendo a libertação deste para os tecidos. Isto é muito importante, por exemplo, em caso
de exercício físico, onde a temperatura corporal aumenta e é necessário um maior
fornecimento de oxigénio.
72
Transporte de Dióxido de Carbono pelo Sangue
Verifica-se que, em condições normais de repouso, 4ml de CO2 são transportados para
os pulmões por cada 100ml de sangue, transporte esse que ocorre com o referido gás sob três
formas: dissolvido (7%), como bicarbonato através de hidratação (70%) e em combinação
com a hemoglobina (23%).
Relativamente ao transporte de CO2 dissolvido no plasma, em condições normais apenas
0.3 ml desse gás são transportados dessa forma por cada 100 ml de sangue.
Para que ocorra o transporte de CO2 sob a forma de ião bicarbonato, este sofre uma
reacção de hidratação em que o CO2 dissolvido no sangue reage com a água, por acção da
anidrase carbónica, que existe e actua no interior dos glóbulos vermelhos, formando ácido
carbónico:
O ácido carbónico formado é imediatamente dissociado em hidrogénio e iões
bicarbonato:
A anidrase carbónica acelera a reacção do CO2 com a água cerca de 5000 vezes. Assim,
esta reacção atinge o equilíbrio em meras fracções de segundo. Esta reacção dá-se ainda antes
de o sangue abandonar os capilares junto ao tecido onde o CO2 foi recolhido. Os iões H+
resultantes da reacção combinam-se com a hemoglobina. Os iões bicarbonato difundem-se
dos glóbulos vermelhos para o plasma. De forma a manter a electroneutralidade, iões cloreto
são transferidos para o interior dos eritrócitos para ocuparem o lugar dos iões que sairam, o
que requer a presença de uma proteína transportadora.
Esta é a forma mais importante de transporte de CO2 no sangue (verifica-se que por
acção de um inibidor da anidrase carbónica, a acetazolamida, a taxa de transporte de CO2 dos
tecidos para os pulmões é tão baixa que a pCO2 nos tecidos passa dos normais 45mmHg para
80mmHg).
Finalmente, o CO2 pode também ser transportado por combinação com a hemoglobina,
reagindo com os radicais amina desta molécula, formando a carbamino-hemoglobina
(carbamino Hb), e proteínas plasmáticas (a sua quantidade no plasma é apenas um quarto da
de hemoglobina, não sendo o transporte efectuado desta forma muito significativo). A
formação da carbamino-hemoglobina é uma reacção reversível com formação de uma ligação
fraca que é facilmente “destruída” para que o CO2 se liberte para os alvéolos pulmonares,
onde a pCO2 é menor que nos capilares.
Há um efeito característico no transporte de CO2 no sangue e que ocorre principalmente
nos pulmões, o efeito de Haldane, onde se verifica que a ligação de O2 à hemoglobina causa
a menor afinidade desta para com as moléculas de CO2 (a hemoglobina torna-se mais ácida).
Verifica-se assim uma dissociação do CO2 da hemoglobina e a libertação do excesso de iões
H+ da hemoglobina (os produtos libertados vão levar ao processo inverso da formação do
ácido carbónico, obtendo-se água e CO2).
Verifica-se também o efeito de Bohr, segundo o qual as altas concentrações de H+ e de
CO2 no sangue causam uma diminuição da afinidade da hemoglobina para com o O2 (o CO2
liga-se a hemoglobina impedindo que o O2 o faça). Em tecidos em rápida metabolização, tais
como o músculo em contracção, muito CO2 e ácido são produzidos. A presença de maiores
níveis de CO2 e H+ nos capilares de tal tecido metabolicamente activo promove a libertação de
O2 da oxi-hemoglobina. Este importante mecanismo para enfrentar a maior necessidade de
oxigénio nos tecidos metabolicamente activos foi descoberto por Christian Bohr, em 1904.
Este efeito verifica-se maioritariamente junto dos tecidos.
Os iões formados a quando do transporte, nomeadamente H+, levam a uma diminuição
do valor do pH do sangue, em parte contrariada pela acção de tampões que diminuem [H+]
(diminuição do pH de 7.41 para 7.37 quando o CO2 entra em circulação).
73
3.7- MÚSCULO E CONTRACÇÃO MUSCULAR
A unidade de organização histológica do músculo esquelético é a fibra muscular, uma célula
larga e cilíndrica, multinucleada. Grupos de fibras musculares agrupam-se formando
fascículos que, finalmente, se associam para formar os diferentes tipos de músculos. O
interior da fibra muscular está ocu
diâmetro. Cada fibra pode conter, desde várias centenas, até muitos milhares de miofibrilhas.
Por sua vez, cada miofibrilha apresenta cerca de 1500 filamentos de miosina e 3000 de
actina, dispostos lado a lado. Em cortes longitudinais pode ser observada a estriação
transversal tão característica das miofibrilhas. Esta estriação é devida à presença de actina e
miosina, as duas principais proteínas contrácteis do músculo. A banda I apresenta-se mais
clara porque a luz polarizada atravessa facilmente os finos filamentos de actina que a
constituem. A banda A apresenta-se mais escura por ser composta por actina e espessos
filamentos de miosina, o que dificulta a passagem da luz. O comprimento relativo das bandas
varia consoante o músculo examinado se encontre em posição de repouso ou contracção. O
comprimento da banda A permanece constante em todas as fases de contracção, mas a banda I
é maior na posição de repouso e menor no músculo contraído. No meio de cada banda I existe
uma linha transversal escura - linha Z, que une miofibrilhas adjacentes. Os filamentos de
actina estão ligados à linha Z, estendendo-se para cada lado dessa membrana para interagirem
com os filamentos de miosina. A unidade estrutural a que se referem todos os fenómenos
morfológicos do ciclo contráctil é o sarcómero, que se define como sendo o segmento
compreendido entre duas linhas Z consecutivas, incluindo uma banda A e a metade de duas
bandas I contíguas. Cada fibra muscular está revestida por uma membrana - sarcolema. Os
núcleos da célula muscular estriada são numerosos e o seu número depende do comprimento
da fibra. O sarcoplasma de uma fibra muscular pode definir-se como o conteúdo do
sarcolema quando se excluem os núcleos. O retículo sarcoplasmático (RS) é um sistema
contínuo de sarcotúbulos limitados por membranas, que se estende por todo o sarcoplasma
formando uma rede tubular de malha fina que envolve cada miofibrilha. Os túbulos
longitudinais distribuem-se em intervalos regulares ao longo das miofibrilhas, confluindo em
canais orientados transversalmente e de calibre maior - cisternas terminais. Pares paralelos
de cisternas terminais distribuem-se transversalmente entre as miofibrilhas ligando-se a um
elemento intermédio de menor diâmetro - túbulo T. Os túbulos terminais longitudinais e as
cisternas terminais do RS estão intimamente relacionados com a libertação dos iões Ca2+.
74
As diferenças observadas nas fibras musculares são resultado da diversidade das proteínas que
as constituem. Os componentes contrácteis básicos da fibra muscular são quatro proteínas
agregadas em dois componentes multimoleculares, os filamentos grossos de miosina e os
filamentos finos de actina, tropomiosina e troponina. Nenhuma proteína por si só apresenta
propriedades contrácteis.
O filamento de miosina é composto por cerca de 300 moléculas de miosina. A
molécula individual de miosina é constituída por seis cadeias polipeptídicas, ou seja, por duas
cadeias pesadas e quatro cadeias leves. Existem dez isoformas diferentes das cadeias pesadas
de miosina. As isoformas têm actividade biológica semelhantes, mas são constituídas por
diferentes aminoácidos. Para cada um dos tipos de cadeias leves de miosina também existem
isoformas (lentas e rápidas). As duas cadeias pesadas formam uma dupla hélice, em que cada
cadeia se apresenta com uma das extremidades enrolada, formando conjuntamente duas
massas de proteína globular, designadas por cabeças de miosina. Deste modo, existem duas
cabeças livres, lado a lado, numa das extremidades da dupla hélice da molécula de miosina.
As cabeças das moléculas de miosina são ainda constituídas pelas quatro cadeias leves (duas
por cabeça), que ajudam no controlo da função das cabeças durante o processo de contracção
muscular. O local responsável pela actividade enzimática da molécula de miosina e pela
afinidade com a actina são as cabeças. Por outro lado é na cauda da molécula de miosina que
se encontram os locais de afinidade desta para com as restantes moléculas adjacentes de
miosina. A cauda é composta pela restante porção em dupla hélice das cadeias pesadas de
miosina. Pelo agrupamento das caudas das moléculas de miosina forma-se o corpo do
filamento de miosina. As cabeças de miosina projectam – se para o exterior deste filamento.
Existe uma pequena parte da porção em dupla hélice de cada molécula de miosina que se
afasta identicamente do corpo do filamento acompanhando a cabeça e constituindo um braço
que possibilita o afastamento das cabeças de miosina relativamente ao corpo do filamento. O
braço e a cabeça de miosina designam-se conjuntamente por ponte transversa (PT).
Várias centenas destas moléculas de miosina encontram-se agrupadas em feixes, com
as cabeças viradas numa direcção ao longo de metade do filamento, e na direcção oposta na
outra metade, deixando uma região média livre e isenta de projecções numa distância de
-se para fora na direcção
dos filamentos de actina e são os únicos elos de ligação, estruturais e mecânicos, entre os
filamentos grossos e finos. O filamento de actina é também um filamento complexo,
composto por três partes distintas: actina, tropomiosina e troponina. Existem várias isoformas
de tropomiosina e de todos os três componentes da troponina, contudo conhece-se apenas uma
forma de actina. A actina é a principal componente deste filamento e as troponina e
tropomiosina são conhecidas como proteínas reguladoras.
75
A parte central do filamento de actina é uma molécula proteica constituída por uma
dupla fita de actina F enrolada em hélice. Cada fita da dupla hélice de actina F é composta de
moléculas polimerizadas de actina G (monómeros). Existem cerca de 13 dessas moléculas por
cada volta, de cada fita, da hélice. A cada uma das moléculas de actina G encontra-se fixa
uma molécula de ADP, constituindo assim os locais activos dos filamentos de actina, com os
quais interagem as pontes transversas dos filamentos de miosina para promoverem a
contracção muscular.
O filamento de actina contém também duas fitas adicionais de proteína que são
polímeros de moléculas de tropomiosina. Pensa-se que cada fita de tropomiosina está
fracamente ligada a uma de actina F. Assim, no estado de repouso, cobre os locais activos da
actina impedindo a interacção actomiosínica e consequentemente a contracção muscular.
Existe também um complexo de três moléculas proteicas globulares, denominado
troponina. Uma dessas proteínas globulares tem grande afinidade pela actina - troponina I,
outra pela tropomiosina - troponina T, e a terceira pelos iões Ca2+ - troponina C. Este
complexo fixa a tropomiosina à actina funcionando como um interruptor, "ligando" ou
"desligando" o filamento de actina. A grande afinidade da troponina pelos iões Ca2+ inicia o
processo de contracção.
Existem outras proteínas adicionais que desempenham diversos papéis na estrutura e
-actinina, desmina e calcioneurina.
A contracção muscular consiste, essencialmente, na ligação e deslizamento das
cabeças de miosina sobre os filamentos de actina F. A ligação da actina à miosina implica
alterações na conformação, que são muito importantes, sobretudo na cabeça de miosina. Estas
alterações dependem do nucleótido presente (ADP ou ATP). As alterações conformacionais
promovem o impulso de força responsável pelo movimento dos filamentos de actina em
relação aos de miosina. A energia envolvida neste processo é fornecida, em última instância,
pela hidrólise do ATP a ADP e Pi. O impulso de força por si próprio ocorre como
consequência de alterações na conformação da cabeça de miosina, quando o ADP se desliga
da mesma.
Os principais eventos bioquímicos durante um ciclo de contracção e relaxamento
muscular podem ser representados por cinco etapas. Na fase de relaxamento da contracção
muscular, a cabeça de miosina hidrolisa ATP a ADP e Pi. Estes compostos permanecem
ligados. O complexo ADP-Pi-miosina resultante é energético e encontra-se por isso numa
conformação de elevada energia. No início desta fase o Ca2+ sarcoplasmático é bombeado
para o RS por ATPases. No interior deste o Ca2+ liga-se a uma proteína específica calsequestrina. Desta ligação resulta uma diminuição da concentração de Ca2+ livre no RS
76
associada a uma diminuição da energia necessária para a remoção destes iões do sarcoplasma
para o RS.
Quando o impulso nervoso que percorre o neurónio motor atinge a junção
neuromuscular, há libertação de acetilcolina (ACH) para a fenda sináptica. Com efeito, a
ACH liga-se a locais específicos (receptores nicotínicos) na membrana da célula muscular,
induzindo uma alteração conformacional na superfície dos canais iónicos. Com a abertura
destes canais apenas os iões de Na+ fluem e despolarizam a membrana da célula.
Paralelamente, ocorre uma propagação desse potencial de acção, através do sistema T, para o
interior da célula. Tal acontecimento provoca a libertação de iões Ca2+ (armazenados nas
cisternas terminais) para o sarcoplasma que contacta com as miofibrilhas. Estes iões
libertados ligam-se então a locais reguladores específicos nas troponinas C presentes nos
filamentos de actina, de facto, a cada molécula de troponina C ligam-se quatro iões Ca2+. Esta
ligação do Ca2+ à troponina provoca uma alteração conformacional no complexo troponinatropomiosina-actina, removendo a inibição mecânica que impedia a interacção entre a actina e
a cabeça da miosina. Portanto, quando a contracção do músculo é estimulada a actina tornase acessível e a cabeça de miosina liga-se a esta. Forma-se então o complexo actina-miosinaADP-Pi. A formação deste complexo promove a libertação de Pi, que inicia o impulso de
força. Segue-se a libertação de ADP. Associada a estes acontecimentos há uma alteração
significativa na conformação da cabeça de miosina relativamente à cauda. A cabeça de
miosina puxa a actina cerca de 10nm na direcção do centro do sarcómero. Estes passos
constituem o impulso de força. A miosina encontra-se agora num estado de baixa energia,
incorporando o complexo actina-miosina. Posteriormente uma outra molécula de ATP liga-se
à cabeça de miosina originando a formação do complexo actina-miosina-ATP. O complexo
miosina-ATP tem pouca afinidade para com a actina, pelo que esta é libertada do complexo
inicial. Esta última etapa é muito importante no processo de relaxamento pois é dependente da
ligação de uma molécula de ATP ao complexo actina-miosina. Começa então um novo ciclo
com a hidrólise de ATP, formando-se de novo a conformação de energia elevada.
A hidrólise de ATP é utilizada como ponto de regulação do ciclo descrito. As regiões
em dobradiça (regiões de flexibilidade da molécula) permitem a grande amplitude do
movimento da cabeça de miosina, bem como o contacto entre as cabeças de miosina e a
actina. Se os níveis intracelulares de ATP diminuírem (por exemplo após morte), este não está
disponível para se ligar à cabeça de miosina e por isto a actina não se dissocia do complexo e
consequentemente não há relaxamento. Esta é a explicação para o rigor mortis.
A contracção muscular depende da energia fornecida pelo ATP. A concentração de
ATP na fibra muscular é apenas suficiente para manter a contracção por 1 a 2 segundos no
máximo. Deste modo, para uma actividade muscular mais intensa é necessário sintetizar ATP.
77
As principais fontes de produção de ATP são a fosfocreatina, glicogénio e respiração celular.
Existem pelo menos dois tipos distintos de fibras no músculo esquelético, uma
predominantemente activa em condições aeróbias (fibras vermelhas) e outra em condições
anaeróbias (fibras brancas). As fibras vermelhas (ricas em mioglobina) utilizam sobretudo a
respiração celular distintamente das fibras brancas que utilizam a fermentação láctica. Ambas
utilizam o ATP preexistente na fibras e as reservas de fosfocreatina.
A fosfocreatina é a fonte a partir da qual a síntese de ATP é mais rápida. Esta contém
ligações fosfato de alta energia. A sua clivagem liberta energia que promove a fixação do ião
fosfato ao ADP, reconstituindo ATP. No entanto, a quantidade de fosfocreatina na fibra
muscular é muito pequena – apenas 5 vezes maior que a de ATP – pelo que a energia
combinada do ATP armazenado com a fosfocreatina existente no músculo só é capaz de
promover uma contracção muscular máxima por apenas 5 a 8 segundos. A segunda fonte de
energia que é usada para reconstituir tanto o ATP como a fosfocreatina é o glicogénio
previamente armazenado no sarcoplasma das células musculares. A libertação de glicose a
partir de glicogénio é dependente de uma glicogénio fosforilase muscular específica, que pode
ser activada por Ca2+, epinefrina e AMP. No sentido de gerar glicose-6-fosfato para a glicólise
no músculo esquelético, a glicogénio fosforilase b deve ser activada em glicogénio
fosforilase, catalisada por uma fosforilase b cinase. O Ca2+ promove a activação desta
fosforilase b cinase também por fosforilação. Assim, o Ca2+ tanto inicia a contracção
muscular como activa uma via para fornecer a energia necessária. O AMP, produzido pela
degradação de ADP durante o exercício muscular, pode também activar a glicogénio
fosforilase b, sem existir fosforilação. A epinefrina também activa a glicogenólise no
músculo. A degradação do glicogénio a piruvato e lactato, liberta energia que será utilizada
para converter o ADP em ATP. Este ATP será usado directamente para “energizar” a
contracção muscular ou para “refazer” as reservas de fosfocreatina. Por outro lado, a
importância do mecanismo da glicólise é dupla: além das reacções glicolíticas poderem
ocorrer na ausência de oxigénio (razão pela qual a contracção muscular pode ser mantida por
muitos segundos mesmo quando não se dispõe de oxigénio), o ritmo de formação de ATP
deste processo é mais rápido que a formação de ATP quando os nutrientes celulares reagem
com o oxigénio. Contudo, acumulam-se muitos produtos terminais da glicólise nas células
musculares de modo que a glicólise é inibida, perdendo-se a capacidade de preservar a
contracção muscular máxima após cerca de 1 minuto. Segue-se a respiração celular (RC), na
presença de oxigénio, ou fermentação do lactato, na ausência de oxigénio. A RC ocorre na
mitocôndria e engloba uma série de reacções envolvendo produção de ATP. As fibras
musculares têm duas fontes de oxigénio: (a) oxigénio que difunde do sangue para as fibras;
(b) oxigénio libertado pela mioglobina no sarcoplasma. A mioglobina é a única proteína que
78
se liga ao oxigénio nas fibras musculares. Na presença de oxigénio, o piruvato entra na
mitocôndria onde é completamente oxidado em reacções que originam ATP, CO2 e H2O. A
RC fornece a maioria do ATP necessário nas actividades mais intensas. Quando os níveis de
oxigénio se encontram baixos como resultado de uma intensa actividade muscular, a maior
parte do piruvato produzido na glicólise é convertido em lactato, num processo designado por
fermentação láctica. Assim a RC envolve maior produção de ATP, cerca de 36 ATP por
cada molécula de glicose relativamente à glicólise anaeróbia.
A contracção prolongada e vigorosa de um músculo leva ao estado conhecido de
fadiga muscular. Um factor importante da fadiga muscular é a redução da libertação de iões
Ca2+ do retículo sarcoplasmático, resultando num declínio de Ca2+ no sarcoplasma. Outros
factores que contribuem para a fadiga muscular incluem o esgotamento da fosfocreatina,
escasso oxigénio, insuficiência de glicogénio e de outros nutrientes, acumulação de lactato e
de ADP, e falha nos impulsos nervosos. Uma vez que o aumento dos níveis de lactato
promovem uma diminuição no pH nos fluidos do corpo, a fadiga muscular pode ser ver vista
como um mecanismo homeostático que impede que o pH caia para valores mais baixos que o
aceitável.
Do ponto de vista anatómico, o músculo liso, é distinto do esquelético por não
apresentar estrias. A actina e a miosina estão presentes e deslizam uma sobre a outra para
produzir a contracção. Em vez das linhas Z, existem corpos densos no citoplasma ligados à
membrana celular e aos filamentos de actina (ligados pela
-actinina). O músculo liso
contém, identicamente ao esquelético, tropomiosina. Contudo a troponina está ausente. Em
geral, as fibras do músculo liso têm poucas mitocôndrias e por isso dependem muito da
glicólise para responder às suas necessidades metabólicas.
O Ca2+ está envolvido no início da contracção do músculo liso, tal como no
esquelético. Uma vez que os retículos endoplasmáticos são pouco desenvolvidos o aumento
da concentração intracelular de Ca2+ que inicia a contracção deve-se principalmente à entrada
do Ca2+ proveniente do líquido extracelular pelos canais de Ca2+ regulados por voltagem.
Além disso, a miosina do músculo liso precisa de ser fosforilada para que possa existir
activação da ATPase da cabeça de miosina. No músculo esquelético também existe
fosforilação e desfosforilação da miosina, contudo a fosforilação não é necessária para a
activação da ATPase. No músculo liso, o Ca2+ liga-se à calmodulina. A calmodulina contém
148 resíduos de aminoácidos e tem quatro domínios de ligação ao Ca2+. Quando a
calmodulina se liga ao Ca2+ adquire a capacidade de activar cinco cinases diferentes. Uma
destas é a cinase da cadeia leve de miosina. As restantes são a fosforilase cinase, as
Ca2+/calmodulina cinases I, II e III. Uma outra proteína também activada pela calmodulina é a
calcineurina. Esta fosfatase inactiva os canais de Ca2+ desfosforilando-os.
79
No músculo liso, da ligação da calmodulina ao Ca2+, resulta um complexo que activa a
cinase da cadeia leve de miosina dependente da calmodulina. Esta enzima cataliza a
fosforilação do resíduo de serina na posição 19 da cadeia leve de miosina. Esta fosforilação
aumenta a actividade da ATPase, distintamente do que ocorre no músculo esquelético e
cardíaco em que a contracção é iniciada pela ligação do Ca2+ à troponina C. A miosina é
desfoforilada pela fosfatase da cadeia leve da miosina da célula. É de notar que a
desfosforilação da cinase da cadeia leve de miosina não resulta necessariamente no
relaxamento do músculo liso porque existem outros mecanismos envolvidos. Um destes é o
mecanismo de ponte trancada, no qual as ligações cruzadas de miosina permanecem ligadas à
actina por algum tempo mesmo depois da diminuição da concentração do Ca2+
sarcoplasmático, permitindo uma contracção sustentada com pouco gasto energético. Esta
contracção sustentada é importante no músculo liso vascular. O relaxamento do músculo
ocorre quando se dá a dissociação total do complexo Ca2+- calmodulina ou quando outro
mecanismo entra em acção. O AMP cíclico participa em reacções que envolvem a
fosforilação da cinase da cadeia leve de miosina. Esta fosforilada exibe uma actividade
significamente menor para com o complexo Ca2+- calmodulina e desta forma é menos
sensível à activação. Assim, um aumento do AMP cíclico inibe a resposta de contracção do
músculo liso, mesmo quando os níveis de Ca2+ intracelular aumentam. Este mecanismo
-adrenérgica. A caldesmona
também desempenha um papel importante na regulação da contracção deste músculo. Em
concentracções reduzidas de Ca2+, a caldesmona liga-se ao complexo tropomiosina-actina,
impedindo a interação actomiosínica e consequentemente a contracção. Para concentracções
elevadas de Ca2+, a caldesmona desliga-se da actina por ligação ao complexo Ca2+calmodulina. Deste modo a actina fica livre para se ligar à miosina permitindo a contracção.
Por outro lado, a caldesmona também esta sujeita a fosforilação/ desfosforilação. Quando
fosforilada não se pode ligar à actina permitindo assim a interacção actomiosínica e
consequentemente a contracção.
As estrias do músculo cardíaco são semelhantes às do músculo esquelético e as linhas
Z também estão presentes. Existem mitocôndrias alongadas em contacto directo com as
miofibrilhas musculares. No ponto em que a extremidade de uma fibra entra em contacto com
outra, as membranas de ambas ficam paralelas, formando uma série extensa de dobras. Estas
áreas são designadas por discos intercalares e localizam-se nas linhas Z. Os discos intercalares
possibilitam uma união firme entre as fibras, mantendo a coesão intracelular permitindo que a
tracção de uma unidade contráctil possa ser transmitida longitudinalmente para a seguinte. O
músculo cardíaco
funciona como se fosse um sincício, mesmo não existindo pontes
protoplasmáticas entre as células.
80
A base molecular de contracção do músculo cardíaco é, de forma geral, semelhante à
do músculo esquelético, uma vez que também nesta estão envolvidas a actina, miosina,
tropomiosina e troponina e o mecanismo é o já descrito anteriormente. O músculo cardíaco,
distintamente do músculo esquelético, exibe ritmicidade intrínseca e miócitos individuais que
se comunicam entre si devido à natureza de sincício. O sistema tubular T é mais desenvolvido
no músculo cardíaco, enquanto o retículo sarcoplasmático é menos extenso. Por esta razão o
fornecimento intracelular de Ca2+ para a contracção é menor. O músculo cardíaco depende,
portanto, do Ca2+ extracelular para a contracção. O AMP cíclico desempenha um papel crucial
no músculo cardíaco. Este controla os níveis intracelulares de Ca2+, através de activações de
cinases. Estas cinases activadas fosforilam várias proteínas de transporte no sarcolema e
retículo sarcoplasmático, assim como do complexo regulador troponina-tropomiosina,
afectando os níveis intracelulares de Ca2+ ou as respostas a ele. Existe uma correlação entre a
fosforilação da troponina I e o aumento da contracção do músculo cardíaco, induzida por
catecolaminas.
Como descrito acima, o Ca2+ extracelular desempenha um papel importante na
contracção do músculo cardíaco. A entrada e saída de Ca2+ é regulada por processos que
envolvem três proteínas transmembranares. Os canais de Ca2+ constituem a principal via de
entrada de iões Ca2+ para o meio intracelular. A principal via de entrada é o canal lento de
Ca2+ regulado por voltagem. Existem também canais rápidos de Ca2+ (presentes no
plasmalema) que, apesar de em menor número, contribuem para o aumento inicial de Ca2+
mioplasmático. Este aumento promove a abertura do canal de libertação de Ca2+ do RS. O
permutador Ca2+- Na+ é a principal via de saída de Ca2+ intracelular. Em repouso contribuí
para a manutenção de um nível baixo de Ca2+ intracelular livre, através da troca de um Ca2+
por três Na+. A energia para o movimento contra gradiente de Ca2+ para fora da célula provém
do movimento a favor do gradiente do Na+ para dentro da célula. Esta troca contribui para o
relaxamento, mas para ocorrer na direção inversa durante a contracção. A Ca2+ ATPase
situada no sarcolema também contribui para a saída destes iões, mas desempenha um papel
relativamente menor quando comparado ao papel do permutador Ca2+- Na+.
81
3.9- Glóbulos Vermelhos
1. Membranas, grupos sanguíneos, composição e funcionalidade
Os glóbulos vermelhos (ou eritrócitos) são as células sanguíneas mais
abundantes, tendo-se num mm3 de sangue, 5 a 6 milhões destas células
num indivíduo do sexo masculino, e 4 a 5 milhões no sexo feminino. Os
glóbulos vermelhos têm a forma de discos bicôncavos, são anucleados e
amitocondriais, apontando-nos este último facto para a ocorrência de
fermentação láctica, para reoxidação do NADH reduzido na glicólise e
produção de ATP, ainda que em regime aeróbio.
A
deformabilidade da estrutura do glóbulo vermelho é uma propriedade
fundamental à passagem deste pelos capilares sanguíneos mais finos, para que não haja
ruptura da membrana. Esta flexibilidade é assegurada pela densa rede citosquelética que
existe no interior da célula, ligada à membrana em diversos pontos. Ao nível da bicamada
fosfolipídica,
podemos
encontrar
proteínas
transmembranares como a banda 3, trocadora de
aniões, aquaporinas, associadas ao movimento de H2O
e as glicoforinas. No citoesqueleto propriamente dito,
surge a espectrina como principal componente desta
rede, formando filamentos que unem os complexos
juncionais. Estes complexos são constituídos por
actina, associada à tropomiosina e à tropomodulina,
que previnem a sua despolimerização. A banda 4.1 e a
aducina, também do complexo juncional, estabilizam
a ligação entre a espectrina e a actina, embora a banda
4.1 promova também a ligação do complexo à
glicoforina na membrana. Tem-se ainda a anquirina,
que vai ligar a própria espectrina a uma banda 3
membranar, facto que sugere uma eficaz interacção
entre o citosqueleto e a membrana, sendo esta não só
ao nível dos complexos juncionais, como também no seio dos filamentos de espectrina
isolados.
Os grupos sanguíneos do sistema AB0 são determinados pela presença ou ausência de
antigénios A e/ou B, na membrana dos glóbulos vermelhos (e de outros tipo de células
sanguíneas). Estes antigénios são oligossacáridos ligados covalentemente a lípidos ou
proteínas, formando glicolípidos ou glicoproteínas. Todas as pessoas possuem a chamada
substância H ou “antigénio” O para alguns autores. Esta substância, sendo produzida por
indivíduos de qualquer tipo sanguíneo, não é na verdade um antigénio, porque não é
susceptível de desencadear uma resposta imunitária. O grupo sanguíneo A vai ser então
identificado pela presença da enzima glicosiltransferase que permite adicionar Nacetilgalactosamina à substância H, para formar o antigénio A. No caso do grupo B, fala-se de
uma glicosiltransferase capaz de adicionar um resíduo de galactose a esta mesma substância
H, produzindo-se um antigénio B. Um indivíduo que seja do grupo AB possui ambas estas
enzimas, logo ambos os antigénios.
No que diz respeito a transfusões sanguíneas, tem-se que o grupo 0 pode
dar sangue a todos os outros grupos, por exemplo, e subentende-se que cada
grupo possa dar a si próprio. Isto deve-se ao facto de se proceder à produção de
anticorpos contra os antigénios inexistentes no grupo em questão. Assim, o
grupo 0 produzirá anticorpos Anti-A e Anti-B, o grupo A produzirá Anti-B, o
grupo B, Anti-A, e o grupo AB, nenhum deles. Assim se justificam as relações
ilustradas na figura ao lado.
No sistema Rhesus, um indivíduo pode ser identificado como Rh + ou Rh
82
– conforme tenha ou não nas membranas dos seus glóbulos vermelhos o antigénio Rh D. Este
antigénio é uma proteína, ao contrário dos do sistema AB0. Um indivíduo Rh – produzirá
anticorpos Anti-D na presença do antigénio, pelo que não pode receber sangue Rh +. No
entanto, o inverso pode suceder.
Um glóbulo vermelho é constituído maioritariamente por hemoglobina, proteína
globular que lhes confere a sua cor característica. É composta por 4 cadeias polipeptídicas
(duas α e duas β), cada uma delas com um grupo prostético Heme. Cada um destes grupos
Heme possui um átomo de ferro no centro, ao qual se pode ligar uma molécula de O2 para o
processo de transporte. Gozando desta característica, os glóbulos vermelhos têm como
principal função o transporte de O2 dos pulmões para os tecidos, sendo que a percentagem de
O2 sanguíneo associado à hemoglobina atinge os 97%. De entre as suas outras
funcionalidades, destaca-se o transporte associado de CO2, aproximadamente 23% do
movimento de CO2, dando-se 70% deste mesmo movimento sob a forma de ião bicarbonato
(HCO3-) dissolvido no plasma. Ainda aqui há intervenção dos glóbulos vermelhos, uma vez
que é a enzima anidrase carbónica, presente nestes, que catalisa a reacção CO2 + H2O ↔ H+ +
HCO3-, ou seja, o CO2 tem de entrar no glóbulo vermelho para se transformar em ião
bicarbonato, que é depois conduzido ao plasma. O H+ pode ser aceite pela parte proteica da
hemoglobina, facto que associa ainda aos glóbulos vermelhos a regulação do pH sanguíneo.
2. Factores de crescimento e produção de glóbulos vermelhos
Os glóbulos vermelhos começam a sua vida na medula óssea a partir de uma célula estaminal
hematopoiética pluripotencial (PHSC), célula essa que pode origem a todos os tipos de células
sanguíneas. Aquando da divisão celular desta, e consequente diferenciação, há uma pequena
porção destas células (PHSC) que se mantêm inalteradas, de modo a garantir a permanência
do processo.
O crescimento e reprodução das diferentes células estaminais são controlados por
múltiplas proteínas, chamadas indutoras ou factores de crescimento. Os factores de
crescimento celular são genericamente divididos em dois subgrupos: citoquinas e
interleucinas. As citoquinas são moléculas moduladoras da proliferação e maturação das
células hematopoiéticas. As interleucinas são proteínas que transmitem sinais de comunicação
entre diferentes tipos de células. A eritropoietina pertence ao subgrupo das citoquinas. A
formação dos factores de crescimento é controlada fora da medula óssea. No caso dos
glóbulos vermelhos, a exposição do sangue a baixas concentrações de oxigénio leva ao seu
aumento em número.
O processo natural de produção de eritrócitos denomina-se
eritropoiese. Especificamente ocorre a partir dos proeritroblastos,
que são grandes células com nucléolos e citoplasma discretamente
disformes. A partir desta célula originam-se por reprodução celular
o eritroblasto basófilo, que após 24/48 horas se transforma por
maturação em eritroblasto policromatófilo. Esta célula vive em
média 24 horas e diferencia-se em eritroblasto ortocromático que 12
horas depois, perde o seu núcleo e dá origem ao reticulócito. O
reticulócito é um eritrócito grande e imaturo, com RNA ribossómico
em varias quantidades no seu citoplasma. Este reticulócito tem um
período de vida médio de 3 dias, após o que se transforma em
eritrócito e é libertado da medula óssea para o sangue circulante. O
eritrócito maduro, por esta altura, contém cerca de 34 % de
hemoglobina (cerca de 90% em peso seco) e não possui organitos,
tendo desde já consigo todas as substâncias de que vai precisar ao
longo da sua vida (120 dias, aproximadamente).
Todo este processo é desencadeado e acelerado
fundamentalmente por uma hormona, um factor de crescimento já referido, a eritropoietina. A
eritropoietina é, quimicamente, uma glicoproteína, é produzida principalmente no rim e, em
83
menor quantidade, no fígado, circulando livremente no sangue. A hipóxia (deficiente
oxigenação sanguínea) é o estímulo principal para a sua produção, qualquer que seja a sua
causa (por exemplo, insuficiência respiratória ou cardíaca, anemia, etc.), é detectada por
sensores de oxigénio nos aparelhos justaglomerulares renais que, como resposta, aumentam a
produção de eritropoietina que, a nível da medula óssea, estimula a síntese e diferenciação de
eritroblastos.
3. Metabolismo do glóbulo vermelho
Antes de mais, é importante referir que os glóbulos vermelhos têm um tempo útil de
vida de aproximadamente 120 dias. Devido ao desgaste na sua membrana plasmática,
provocado pela sua circulação ininterrupta ao longo dos vasos, tornam-se ineficazes, dando-se
a sua destruição essencialmente no baço, mas também no fígado e na medula óssea vermelha.
Macrófagos, nesses locais, fagocitam esses glóbulos vermelhos velhos e fragilizados (e/ou
com rupturas), evitando um grande número de doenças.
Os glóbulos vermelhos sofrem fagocitose por acção dos macrófagos e, através das
enzimas lisossómicas existentes nos vacúolos dos ditos macrófagos, os componentes
químicos dos glóbulos vermelhos separam-se. A hemoglobina é, então, separada em globina e
grupo Heme, seus componentes. A globina é degradada nos seus aminoácidos constituintes,
podendo estes seguir duas vias: ser reutilizados, sendo “aproveitados” para a síntese proteica,
ou ser libertados na corrente sanguínea.
O ferro é removido, nas células fagocitárias, do grupo Heme. Quando não é
armazenado pelos macrófagos, o ferro é libertado na corrente sanguínea podendo, no plasma,
conjugar-se com a proteína transportadora transferrina, que o transporta até à medula
vermelha. Uma vez lá, esse ferro é utilizado, pelas células precursoras de glóbulos vermelhos,
na síntese de hemoglobina. O ferro em excesso pode ser armazenado na medula óssea e no
fígado, sendo que algum é também “perdido” na bílis, sendo que essa é uma das razões pelas
quais devemos incluir sempre alimentos ricos em ferro na nossa alimentação.
A vitamina B12 é também usada na síntese de hemoglobina. O factor intrínseco,
glicoproteína produzida pelas células parietais do estômago, é fundamental na absorção da
Vitamina B12. Os aminoácidos, quando na medula óssea, são utilizados para sintetizar a
porção globina da hemoglobina;
A eritropoietina, hormona produzida pelos rins, e cuja produção é aumentada em
situações de hipóxia (deficiência de oxigénio), circula através do sangue até à medula óssea
onde vai estimular a eritropoiese.
A eritropoiese e a degradação dos glóbulos vermelhos procedem, normalmente, ao
mesmo ritmo. Se a capacidade de transporte de oxigénio decresce, devido à eritropoiese não
acompanhar o ritmo de degradação dos glóbulos vermelhos, a produção destes aumenta
(através do mecanismo de Feedback negativo já explicado).
Quando o ferro é removido do grupo heme, a parte não ferrosa deste é reduzida a
biliverdina (pigmento verde). A biliverdina, por acção da biliverdina redutase, é de seguida
reduzida a bilirrubina (pigmento amarelo-alaranjado). Essa bilirrubina recém-formada circula,
na forma não conjugada, no sangue ligada à albumina sérica, sendo transportada pelo sistema
porta até o fígado onde, após se conjugar com ácido glicurónico, tornando-se na forma
conjugada (solúvel em água), é secretada para a bílis.
A bilirrubina conjugada, presente na bílis, vai para o intestino delgado e, de seguida,
para o intestino grosso onde, após lhe ser removido o ácido glucurónico, se torna, por acção
de bactérias, em urobilinogénio. Uma pequena porção do urobilinogénio formado é
reabsorvido pelo intestino e, seguidamente, excretado pelo rim onde é oxidado a urobilina
(pigmento amarelo que dá a cor à urina), ou seguindo de novo para o fígado. Mas a grande
maioria do urobilinogénio não é reabsorvido, permanecendo no intestino grosso onde é
oxidado a estercobilina (pigmento castanho), sendo desta forma eliminado nas fezes (cuja cor
acastanhada advém da estercobilina).
84
Lipidémia
a) Formação e destinos metabólicos das lipoproteínas
Os lípidos são transportados no plasma na forma de lipoproteínas.
As 4 maiores classes de lípidos estão presentes nas lipoproteínas são: triacilgliceróis,
fosfolípidos, colesterol, “cholesteryl esters” e uma fracção muito pequena de ácidos
gordos livres (é a forma mais activa dos lípidos plasmáticos). As quantidades de cada um
destes lípidos varia de lipoproteína para lipoproteína.
Os 4 maiores grupos de lipoproteínas são:
- Quilomicra: são formadas no intestino. Derivam da absorção intestinal dos
triacilgliceróis ou outros lípidos. São as lipoproteínas menos densas.
- VLDL (very low density lipoproteins): formadas no fígado. Lípidos predominantes:
triacilgliceróis.
- IDL (intermediate density lipoproteins): derivam do VLDL (também são chamadas
VLDL remanescente). Depois de formado, é convertido em LDL. Lípidos predominantes:
triacilgliceróis.
- LDL (low density lipoproteins): correspondem à fase final do catabolismo das VLDL.
Lípidos predominantes: colesterol.
- HDL (high density lipoproteins): são formadas no fígado e no intestino. Estão
envolvidas no metabolismo dos Quilomicra e das VLDL e também no transporte de
colesterol. São as lipoproteínas mais densas. A sua função principal é mobilizarem o
excesso de colesterol dos tecidos para o fígado, onde é armazenado. Lípidos
predominantes: fosfolípidos e colesterol.
Estrutura das lipoproteínas:
As lipoproteínas são formadas por um núcleo apolar
(constituído
principalmente
por
triacilgliceróis
e
“cholesteryl esters”), uma única camada superficial de
lípidos anfipáticos (fosfolípidos e moléculas de colesterol
livre) e uma parte proteica. Têm uma forma
aproximadamente esférica, devido, precisamente, à sua
estrutura (com os grupos hidrofílicos na superfície e os
grupos hidrofóbicos no interior).
A parte proteica da lipoproteína é chamada
apoproteína. É a distribuição das apoproteínas que
caracteriza a lipoproteína. Em cada lipoproteína pode estar
presente uma ou mais apoproteínas. A quantidade desta
parte proteica presente em cada lipoproteína é muito
diferente; por exemplo, nas HDL constitui cerca de 70% da molécula, enquanto que nos
quilomicra constitui apenas 1%. As funções das apoproteínas são: serem cofactores
enzimáticos ou inibidores enzimáticos; fazerem a ligação entre a lipoproteína e os
receptores lipoproteicos nos tecidos.
Enzimas e proteínas de transporte:
A maioria dos sistemas enzimáticos participantes no metabolismo lipoproteico
participa na hidrólise das lipoproteínas ricas em triacilglicerol após as refeições e têm uma
diminuta actividade durante o jejum.
lipoproteína lipase (LpL): sintetizada pelos adipócitos, miócitos (no músculo
cardíaco e esquelético) e macrófagos; activada pela apoC-II (e fosfolípidos); determina a
hidrólise do triacilglicerol (perda de 90%), o que leva à libertação de ácidos gordos para
os tecidos (utilizados como fonte de energia nos músculos ou na síntese hepática de
VLDL e acumulados, como triacilglicerol, no tecido adiposo); origina a perda da apo C
dos quilomicra e das VLDL; interactua com quilomicra e VLDL em circulação: leva à
85
formação de quilomicra e VLDL remanescentes, que são ricos em colesterol e
“cholesteryl esters” (devido à perda de triacilglicerol).
lipase hepática (LH): sintetizada pelos hepatócitos; relacionada com o processamento
final dos remanescentes de quilomicra; promove a hidrólise do triacilglicerol e dos
fosfolípidos em excesso na sua membrana; favorece a interacção das lipoproteínas
remanescentes com o receptor LRP; acelera o metabolismo das IDL até LDL.
lecitina-colesterol aciltransferase (LCAT): sintetizada no fígado; circula no plasma
associada às HDL (que são o seu principal substrato); catalisa a esterificação do colesterol
livre, ao promover a transferência de ácidos gordos da lecitina para o colesterol (com
formação de ésteres de colesterl e lisofosfatidilcolina); cofactor: apoA-I, mas outras
apoproteínas (AII, A-IV, C-I, D e E) podem também activar a enzima.
proteína de transferência dos “cholesteryl esters” (CETP): não é uma enzima, mas sim
uma proteína produzida pelo fígado; promove a transferência de “cholesteryl esters” das
HDL para as VLDL, IDL e remanescentes e de triacilglicerol em sentido inverso;
juntamente com a LCAT, a CETP está envolvida no metabolismo das HDL.
Receptores celulares envolvidos no metabolismo lipoproteico:
receptor LDL (LDLR): glicoproteína constituída por cinco domínios funcionais;
expresso na maioria das células com núcleo (em especial no fígado); envolvido na
captação das lipoproteínas com apo B e/ou apo E (LDL, Quilomicra remanescente, IDL e
HDL1) e, consequentemente, na regulação do conteúdo intracelular de colesterol.
LRP (LDLreceptor related protein): receptor de membrana multifuncional;
estruturalmente comparável a quatro receptores LDL (com 31 domínios funcionais);
expresso, primariamente, no fígado, mas também no cérebro (neurónios) e na placenta;
envolvido na captação hepática de remanescentes (de Quilomicra e VLDL) ricos em apo
E.
receptor VLDL: estruturalmente semelhante ao LDLR (com oito domínios funcionais);
abundante nos hepatócitos, adipócitos e SNC; capaz de fixar lipoproteínas com apoE.
receptores HDL: papel central no transporte reverso do colesterol; presente nos
hepatócitos; medeia a captação selectiva de colesterol e “cholesteryl esters” das HDL,
contribuindo de forma importante para a homeostasia global do colesterol no organismo;
presente também nas células produtoras de esteróides onde facilita a captação do
colesterol necessário à síntese hormonal.
Concluído a apresentação dos diversos elementos presentes, podemos mais facilmente
compreender a formação de lipoproteínas e o metabolismo lipoproteico.
Formação de Quilomicra e VLDL
Os quilomicra são produzidas pelas células intestinais. As VLDL são produzidas nas
células hepáticas. O mecanismo de formação dos quilomicra e das VLDL é semelhante.
Os quilomicra encontram-se no quilo. São formadas apenas pelo sistema linfático que
drena o intestino. São responsáveis pelo transporte de todos os lípidos na circulação.
As VLDL são o meio de transporte de triacilglicerol desde o fígado até aos tecidos
extrahepáticos.
Quando os quilomicra e as VLDL são formadas contém apenas uma pequena
quantidade de apoproteínas C e E; o complemento todo é obtido a partir das HDL na
circulação.
A apo B é essencial para a formação tanto dos quilomicra como das VLDL.
Tanto no caso dos quilomicra como no caso das VLDL, a apo B é sintetizada no
retículo endoplasmático rugoso. Posteriormente, é incorporada nas lipoproteínas no
retículo endoplasmático liso, que é o principal local da síntese de triacilglicerol. Depois da
adição de resíduos de hidratos de carbono no complexo de Golgi, os quilomicra são
libertadas para o sistema linfático. Quanto às VLDL, são secretadas dentro do espaço de
Disse (espaço entre o hepatócito e o sinusóide, que contém o plasma sanguíneo) e depois
dentro dos sinusóides hepáticos (pequenos vasos sanguíneos semelhantes a capilares mas
86
que contém endotélio fenestrado), sendo depois libertadas no lúmen dos capilares
sanguíneos.
O triacilglicerol é transportado a partir do intestino nos quilomicra e a partir do fígado
nas VLDL.
Metabolismo lipoproteico
A principal função do metabolismo lipoproteico é o transporte dos triacilglicerol e
colesterol do intestino e do fígado para os locais de reserva e utilização metabólica.
Por isso, globalmente, pode ser entendido como 3 vias:
- via exógena, que compreende a absorção e o transporte das gorduras da dieta até ao fígado
(e aos tecidos);
- via endógena, que abarca o transporte (e o metabolismo) das VLDL produzidas no fígado;
- transporte reverso do colesterol, que está relacionada com a condução do colesterol,
possivelmente em excesso, dos tecidos periféricos para o fígado.
(1) Via exógena (Metabolismo dos quilomicra)
As gorduras alimentares, depois
de emulsionadas e misturadas com os
sais biliares e com as lipases
pancreáticas, são absorvidas no
jejuno. Nos enterócitos, os ácidos
gordos e o colesterol derivados da
dieta são esterificados, de modo a
reconstituir o triacilglicerol e a
formar, conjuntamente com a apoB48, sintetizada no intestino, os
quilomicra nascentes, que entram na
circulação (vindas do sistema
linfático).
Durante
a
passagem
pela
circulação sistémica os quilomicra
adquirem, das HDL, apoE e apoC-I, C-II e C-III, constituindo os quilomicra propriamente
ditos. Depois, os triacilgliceróis são hidrolisados, pela LpL em ácidos gordos livres, que
são depois armazenados nos tecidos periféricos (de novo, sob a forma de triacilglicerol),
utilizados como fonte energética ou reutilizados na síntese de outras lipoproteínas pelo
fígado. Deste processo resultam os quilomicra remanescentes, mais pobres em
triacilglicerol e mais ricas em colesterol e já sem a apo A e apo C (que regressam às HDL
nascentes, contribuindo assim para a permanente reciclagem e remodelagem das
lipoproteínas no plasma). Depois disto, os quilomicra vão para o fígado (onde se ligam ao
LRP ou ao receptor HDL), onde são convertidas em ácidos gordos e colesterol.
(2) Via endógena (Metabolismo das VLDL e LDL)
As VLDL, sintetizadas no fígado a
partir dos triacilgliceróis e fosfolípidos,
entram na circulação e sofrem um
processo semelhante ao descrito para os
quilomicra.
Desde a sua constituição – ou
adquiridas na circulação – as VLDL têm
apoB-100, apoE e várias formas de apoC,
importantes para a modulação do seu
metabolismo (por exemplo, a apoE é o
ligando crítico de reconhecimento pelos
receptores LRP e LDL, enquanto que
87
apoC-II é um elemento central no metabolismo das VLDL ao co-activar a LpL).
Na circulação, as VLDL são hidrolisadas pela LpL (e também pela lipase hepática do
triacilglicerol) e são progressivamente convertidas em partículas cada vez mais pequenas e
mais ricas em colesterol (as VLDL remanescentes ou IDL), que podem seguir 2 vias: ser
depuradas pelo fígado (via receptor LDL) ou ser convertidas em LDL.
As LDL resultantes apenas contêm colesterol e a apo B-100. Vão, então, ser captadas pelo
fígado e pelos tecidos periféricos, via receptor LDL (LDLR).
(3) Transporte reverso do colesterol (Metabolismo das HDL)
As HDL são um conjunto heterogéneo de lipoproteínas, diversas do ponto de vista
estrutural e funcional. Podem ter origens diferentes: serem segregadas pelo fígado,
sintetizadas directamente pelo intestino (num e noutro caso sob a forma de HDL nascentes ou
HDL3), derivarem dos “restos” redundantes do catabolismo das lipoproteínas ricas em
triacilglicerol ou resultarem da interconversão das HDL2 e HDL3 – pela acção da CETP, da
PLTP ou da lipase hepática.
As HDL nascentes têm uma forma
de disco, constituído por fosfolípidos e
colesterol, encontrando-se ligada a apo
A-1 e a enzima LCAT. Esta enzima
permite a conversão do colesterol em
“cholesteryl esters” e lisolecitina. Ao
mesmo tempo, a molécula de HDL
recebe dos tecidos colesterol, levando à
formação da HDL3, que tem uma
forma esférica, com os lípidos apolares
(“cholesteryl esters”) no interior e os
lípidos polares (colesterol e
fosfolípidos) no exterior e contendo
ainda a apo A-1 ligada e a enzima
LCAT, que vai novamente converter o colesterol em “cholesteryl esters” e lisolecitina. Ao
mesmo tempo, a HDL3 vai receber dos tecidos (através do receptor ABC-1 (ATP-binding
cassette transporter-1) mais colesterol, levando à formação de uma molécula com maiores
dimensões, a HDL2, que já não tem a LCAT ligada. Depois disto, a HDL2 pode seguir 2 vias:
ser reconvertida a HDL3 ou ser destruída. Nesta última via, a apo A-1 separa-se da
lipoproteína e pode ir directamente para o rim (onde é destruída) ou incorporar a Preβ-HDL,
que é a forma mais potente de HDL; enquanto isso, os “cholesteryl esters” vão-se ligar ao
receptor SR-B1 (scavenger receptor class B1), no fígado. O colesterol e os fosfolípidos que
constituíam a HDL2 também vão para o fígado.
b) Formação e mobilização dos depósitos lipídicos
Grandes quantidades de lípidos podem ser armazenadas no tecido adiposo. Além do
armazenamento de triacilgliceróis, o tecido adiposo desempenha funções como isolante
térmico e de protecção mecânica.
Formação de depósitos lipídicos
Na formação dos depósitos lipídicos encontramos 2 possíveis fontes de triacilgliceróis:
fígado (é a principal); alimentação (engloba os lípidos absorvidos ao nível do intestino e a
glicose que possibilita a síntese dos triacilgliceróis no tecido adiposo).
Os triacilgliceróis são formados a partir da esterificação de acil-CoA e glicerol 3-fosfato. O
glicerol 3-fosfato utilizado nesta esterificação, e uma vez que no tecido adiposo não se
encontra presente a enzima glicerol cinase, é sintetizado a partir da glicose, via glicólise,
sendo o resultado da redução da dihidroxiacetona fosfato. A enzima envolvida na reacção de
redução é a glicerol 3-fosfato desidrogenase. A acil-CoA pode ter 3 proveniências:
88
Os quilomicra (provenientes do intestino) e as VLDL (provenientes do fígado)
sofrem a acção de uma enzima, a lipoproteína lipase, que se encontra na face luminal
da membrana das células endoteliais dos capilares do tecido adiposo, que catalisa a
hidrólise dos triacilgliceróis em três ácidos gordos e glicerol. Os ácidos gordos livres
resultantes entram na sua maioria para os adipócitos e, por acção da acil-CoA
sintetase, na presença de CoA e ATP ( por cada ácido gordo activado gasta-se um
ATP), dão origem a acil-CoA. Alguns destes ácidos gordos podem não entrar nos
adipócitos, ficando na corrente sanguínea e sendo transportados em conjunto com a
albumina.
A glicose que entra para os adipócitos pode ter vários destinos metabólicos: via das
fosfopentoses, sendo oxidada a CO2; via glicólise, originando acetil-CoA; ciclo de
Krebs, importante na produção de NADPH, que é utilizado na obtenção de acil-CoA,
a partir de acetil-CoA (glicólise).
Os ácidos gordos provenientes da lipólise podem seguir 2 vias: circulação sanguínea,
sendo transportados com a albumina ou podem dar origem novamente a acil-CoA,
através da acil-CoA sintetase.
A esterificação dos triacilgliceróis ocorre em 2 fases. Na primeira fase:
Remoção dos dois primeiros grupos hidroxilo livres do glicerol 3-fosfato e
adição de 2 ácidos gordos nesses pontos. Esta reacção é catalisada pela acilCoA sintetase, na presença da acil-transferase;
A molécula resultante desta fase é o ácido fosfatídico;
Numa segunda fase:
O grupo fosfato do ácido fosfatídico é hidrolisado pela fosfatidato
fosfatase, formando o 1,2-diacilglicerol;
O diacilglicerol é transesterificado com um terceiro ácido gordo, através da
aciltransferase, originando o triacilglicerol.
No citoplasma dos adipócitos existe (em todos os estados metabólicos) um ciclo de hidrólise
(catalisada pela lípase hormono-sensível) e síntese (esterificação) de triacilgliceróis.
No tecido adiposo, a seguir às refeições a actividade da lípase de lipoproteínas está,
relativamente ao estado de jejum, aumentada promovendo a hidrólise dos triacilgliceróis dos
quilomicra. A activação da lípase de lipoproteínas deve-se, sobretudo, à acção da insulina que
estimula a sua síntese nos adipócitos e a sua migração para o endotélio. Além desta acção, a
insulina inibe a lipólise e favorece o processo de esterificação, estimulando a síntese de aciltransferase do glicerol 3-fosfato, a primeira enzima no processo da síntese dos triacilgliceróis.
Os quilomicra, via activação da síntese e libertação de uma citocina dos adipócitos também
estimulam a síntese de triacilgliceróis, nos adipócitos.
O consumo dos ácidos gordos na esterificação favorece a sua captação para dentro dos
adipócitos. No período que se segue a uma refeição o processo de captação de ácidos gordos e
esterificação é, portanto, mais rápido que o de hidrólise, ocorrendo acumulação de gordura
nos adipócitos.
Mobilização dos depósitos lipídicos
A lipólise (degradação) dos triacilgliceróis, nos adipócitos, é mediada pela lipase hormonosensível (inibida pela insulina e estimulada pelas catecolaminas), que promove a libertação
dos três ácidos gordos e do glicerol. Os ácidos gordos resultantes podem contribuir
novamente para a formação de triacilgliceróis, ou então ser libertados para a corrente
sanguínea, onde são transportados conjuntamente com a albumina, com destino a tecidos
consumidores como o músculo e o fígado, onde os ácidos gordos se separam da albumina
para entrarem nas células e sofrerem β-oxidação. O glicerol resultante vai para a circulação e
segue para tecidos, como o fígado ou rins, onde está presente a enzima glicerol cinase que
permite a regeneração da glicose, via gliconeogénese.
Em condições de necessidades energéticas (por exemplo em situações de jejum ou
exercício físico), o ciclo de lipólise-esterificação no tecido adiposo também ocorre mas, como
89
a lipólise é mais rápida que a esterificação, o somatório dos dois processos corresponde à
libertação de ácidos gordos livres dos adipócitos para o plasma. No jejum não há quilomicra,
a insulina plasmática está baixa e os adipócitos têm maior sensibilidade à acção lipolítica das
catecolaminas. Nestas condições, os ácidos gordos livres estão aumentados no plasma e são
usados como “combustíveis” pelos tecidos nomeadamente os músculos e o fígado.
c) Controlo metabólico da lipidémia e utilização metabólica tecidual dos
lípidos
As reservas de triacilgliceróis no tecido adiposo estão constantemente a sofrer lipólise e
reesterificação. Estes dois processos são vias completamente distintas, envolvendo diferentes
substratos e enzimas. Isto permite que os dois processos sejam regulados separadamente por
diversos factores nutricionais, metabólicos e hormonais. O resultante destes dois processos
determina a quantidade de ácidos gordos livres presentes no tecido adiposo, e
consequentemente, a concentração destes em circulação no plasma.
Triacilglicerol é formado a partir da esterificação de acil-CoA e glicerol-3-fosfato.
Dado que no tecido adiposo não está presente a enzima glicerol cinase, o glicerol-3-fosfato
tem de ser sintetizado a partir da glicólise, mais concretamente da redução dihidroxiacetona
fosfato.
No entanto, a glicose que entra no tecido adiposo tem outros destinos metabólicos, tais
como a oxidação a CO2, na via das fosfopentoses ou ciclo de Krebs, e produção de NADPH,
que é essencial na produção de acil-CoA partindo de acetil-CoA (obtido através da glicólise).
Assim, um aumento da utilização metabólica da glicose (i.e. necessidades energéticas),
vai-se traduzir numa diminuição da libertação de ácidos gordos para a circulação. Isto porque
a glicose vai ser preferencialmente oxidada a CO2, fazendo com que se forme menos
quantidade de glicerol 3-fosfato, o que implica uma menor taxa de esterificação. Os ácidos
gordos formados por lipólise de triacilgliceróis são utilizados para regenerar acil-CoA (devido
à enzima acil-CoA sintase), e este dá origem a acetil-CoA que posteriormente entra no ciclo
de Krebs, sendo oxidado a CO2. Deste modo verifica-se uma diminuição de ácidos gordos
livres no adipócito, o que se traduz numa diminuição da lipidémia. É importante referir que
nesta situação, a taxa de libertação de glicerol (formado a partir da hidrólise de
triacilgliceróis) para a circulação se mantém constante, pelo que é possível inferir que a
glicose não actua por diminuição da taxa de lipólise.
Outro aspecto a referir em relação ao efeito da glicose sobre a lipidémia, é que em
situações de jejum verifica-se uma diminuição da glicémia e consequentemente existe uma
diminuição da taxa de esterificação e formação de triacilgliceróis. Deste modo constata-se
uma acumulação de ácidos gordos no adipócito, e consequentemente, um aumento da
concentração destes em circulação. Por outro lado, um aumento de glicose no sangue traduzse num aumento da taxa de esterificação, o que leva à acumulação de triacilgliceróis no
interior do adipócito.
A taxa de libertação de ácidos gordos livres do tecido adiposo é afectada por diversas
hormonas que influenciam, tanto a taxa de esterificação como a de lipólise.
A insulina inibe a libertação de ácidos gordos dos adipócitos, o que se traduz numa
diminuição da lipidémia. Ela aumenta a lipogénese e síntese de acilglicerol, e também a
oxidação da glicose a CO2 pela via das fosfopentoses. Todos estes efeitos estão dependentes
da presença de glicose e podem ser explicados pelo facto de a insulina aumentar o fluxo de
glicose para o adipócito, através do transportador GLUT 4. No entanto, a principal acção da
insulina no tecido adiposo é inibir a actividade da lipase hormono-sensivel, reduzindo a taxa
de lipólise e consequentemente a libertação de ácidos gordos livres e glicerol. Verifica-se
ainda que a insulina diminui a taxa de lipólise por outras vias. Uma delas é inibir a actividade
da adenilato-ciclase, que é responsável pela conversão de ATP em cAMP, que se traduz numa
diminuição da concentração de cAMP na célula. Dado que a lipase hormono-sensível passa da
sua forma inactiva para a forma activa através de uma fosforilação catalisada por uma
proteína cinase dependente de cAMP, uma diminuição deste causa uma menor quantidade de
lipase activa, diminuindo assim a taxa de lipólise. Outra via é estimular a enzima
90
fosfodiesterase, responsável pela conversão de cAMP em 5’ AMP. Uma maior actividade
desta enzima implica uma menor concentração de cAMP, que se traduz numa diminuição da
taxa de lipólise pelo processo acima descrito. A insulina activa ainda a enzima lipase
fosfatase, que é responsável pela conversão da lipase hormono-sensivel da sua forma activa,
para a inactiva, através de uma desfosforilação. Isto faz com que haja menos lipase activa e
consequentemente, menor taxa de lipólise. Todas estas vias de diminuição da libertação de
ácidos gordos para a circulação, são vias directas. Além de actuar sobre estas, a insulina inibe
ainda uma via indirecta independente de cAMP, a via de produção da lipase hormonosensivel. Isto faz com que haja uma diminuição da síntese de lipase e consequentemente, uma
menor taxa de lipólise.
Outras hormonas responsáveis pela diminuição da libertação de ácidos gordos livres
são a prostaglandina E1 e ácido nicotínico, que inibem a adenilato-ciclase diminuindo a
lipólise pelo processo já descrito (actuam sobre a adenilato-ciclase de forma análoga à
insulina).
Outras hormonas aceleram a libertação de ácidos gordos livres do tecido adiposo,
aumentando a taxa de lipólise e aumentando assim a lipidémia. As que actuam rapidamente
na promoção de lipólise incluem as catecolaminas (epinefrina e norepinefrina), ACTH, TSH,
GH, e actuam por estimulação da adenilato-ciclase, aumentando a conversão de ATP em
cAMP, e consequentemente, a quantidade de lipase hormono-sensível activa, aumentando a
degradação de triacilgliceróis a ácidos gordos e glicerol. As metilxantinas (i.e. cafeína) inibem
a actividade da enzima fosfodiesterase, causando uma diminuição na conversão de cAMP em
5’ AMP e portanto, mantendo a taxa de lipólise elevada. Os glicocorticoides (que são
hormonas esteroides e portanto derivadas do colesterol), difundem facilmente através da
membrana celular dos adipócitos, ligando-se a receptores específicos do núcleo, aumentando
a síntese de lipase hormono-sensivel, por uma via independente de cAMP. Todas estas
hormonas têm efeitos antagónicos à insulina.
Falando agora da utilização metabólica dos lípidos. Já foi falado que os triacilgliceróis
são transportados em circulção, incorporados em lipoprotéinas (quilomicra e VLDL), que
após a acção da lipoproteína lipase libertam ácidos gordos e glicerol. Os ácidos gordos entram
nos tecidos através de uma proteína transportadora que actua por co-trasnporte mediado pelo
Na+. Consoante o tecido, os ácidos gordos terão diferentes aplicações. No caso do músculo
(esquelético e cardíaco), os ácidos gordos sofrem principalmente β-oxidação, visto serem a
principal fonte de energia destes tecidos em situação de jejum. No caso do tecido adiposo, os
ácidos gordos são utilizados principalmente para reesterificação de triacilgliceróis, através da
síntese de acil-CoA. É importante referir que o músculo tem muito maior afinidade para os
ácidos gordos do que o tecido adiposo, captando praticamente a totalidade libertada pela
acção da LPL, enquanto que o tecido adiposo apenas capta cerca de 2/3. Os ácidos gordos
restantes ligam-se à albumina e são transportados para outros tecidos, nomeadamente o
fígado. Esta diferença de afinidade é importante em situações de jejum, fazendo com que o
músculo (principalmente o cardíaco) tenha assegurada a quantidade necessária de ácidos
gordos para o seu funcionamento.
Outras duas classes de lípidos com elevada importância metabólica são o colesterol e
os fosfolípidos. Embora o colesterol possa ser sintetizado por quase todos os tecidos, a sua
principal fonte é a alimentação. É um elemento essencial nas membranas celulares,
conferindo-lhes fluidez. É também o precursor das hormonas esteróides e vitamina D, que
desempenham um papel fundamental no nosso organismo, actuando das mais diversas
formas. Os fosfolípidos, devido a serem moléculas anfipáticas são os principais constituintes
das membranas celulares (bicamada fosfolipídica). São essenciais nas lipoproteínas, formando
uma monocamada, permitindo a formação de um núcleo hidrofóbico, fundamental no
transporte de lípidos em circulação. É de referir que alguns fosfolípidos de membrana contêm
o ácido araquidónico, libertando-o por acção da fosfolipase A2, em resposta a sinais
hormonais (a fosfolipase A2 cliva a ligação éster entre o ácido araquidónico e o glicerol). O
ácido araquidónico é o precursor dos eicosanóides, que desempenham um papel essencial no
nosso metabolismo.
91
3.11- Glicémia
Formação e mobilização da glicose e glicogénio: “nos hepatocitos” vs “no
músculo”
O fígado é o órgão fundamental na regulação da glicémia (concentração de glicose no
sangue), tendo assim a função de “glicostato”: mantém a glicémia numa gama de valores
normal, sendo esta nos mamíferos de 4.5-5.5 mmol/L.
O mecanismo principal da homeostase da glicose será então a troca entre processos de
utilização (glicólise), formação de reservas (glicogénese) e formação de nova glicose a partir
de substratos não glicídicos (gliconeogénese).
Após ingestão, as moléculas de glicose absorvidas no intestino chegam ao fígado pela
veia porta e são captadas pelos hepatocitos por difusão facilitada, uma vez que não são
capazes de se difundirem simplesmente pela membrana. Para isso, utilizam o transportador
predominante GLUT-2.
Como possíveis destinos da glicose, vamos ter então:
- Degradação para fornecimento de energia de células (glicólise, ciclo de krebs).
- Síntese de glicogénio para reserva (até 10% do peso total do fígado) pela
glicogénese.
- Em excesso, também para reserva, formação de triacilgliceróis, a partir de acetilCoA.
- Via das fosfopentoses, para formação de transportadores (NADPH) e nucleótidos.
Visão Geral dos processos envolvidos:
Glicólise: como já vimos, com o intuito de fornecer energia às células, mas também para
fornecer esqueletos de carbono para vias de síntese. De notar que ao degradar sacáridos com
pelo menos duas unidades, temos de transformar todo os monossacáridos resultantes em
glicose para prosseguir a via. Isso faz-se através de uma série de reacções, em que intervêm
enzimas como uridiltransferase, epimerases e o intermediário UDP-Glicose para finalmente
formar a Glicose-1-fostato. Note-se que a passagem de glicose a glicose-6-fosfato é éfectuada
no fígado pela hexocinase IV enquanto no músculo as enzimas intervenientes são a
hexocinase I e II.
Gliconeogénese: para formar então a nova glicose, a partir de percursores como: piruvato,
lactato, glicerol e produtos do catabolismo de alguns aminoácidos, que entram no ciclo em
pontos diferentes. A glicose produzida nos hepatócitos é então exportada para outros tecidos
quando as reservas de glicogénio estão (praticamente) esgotadas. Em estado recémalimentado poderá conduzir também à síntese de glicogénio. Para exportar então a glicose,
temos o transporte da Glicose-6-fosfato do citosol para o lúmen do retículo endoplasmático
do hepatocito (pelo transportador T1). Aí, por acção da enzima glicose-6-fosfatase (enzima
esta inexistente no músculo), temos então a molécula de glicose, lançada novamente para o
citosol, junto com o fosfato restante (respectivamente pelos transportadores T2 e T3), e
finalmente, pelo GLUT-2, a glicose é lançada na corrente sanguínea, aumentando a glicémia.
Comparando as duas vias, verifica-se que são o inverso uma da outra (em termos de sentido,
porque as enzimas actuantes são outras, como se sabe), e portanto conclui-se que quando a
glicólise está muito activada, a gliconeogénese está muito inibida.
O glicogénio representa uma menor percentagem do músculo do que representa no
fígado, mas devido à maior massa do músculo, é aí que se encontra a sua maioria, servindo
como “combustível”, como fonte de reserva de glicose para as próprias células musculares.
Isto deve-se ao facto das células musculares não terem a glicose-6-fosfatase, ou seja tendo
incapacidade de passar a glicose para o sangue.
Para os processo envolvidos na formação e mobiliazação do glicogénio, temos:
92
Glicogénese: síntese do glicogénio (reserva), a partir de moléculas de glicose (a enzima
fundamental é a glicogénio sintase). A glicose é transformada em glicose-6-fosfato pela
hexocinase correspondente e a glicose-6-fostato, por uma glicomutase, forma a glicose-1fosfato, que por sua vez é transformada em UDP-glicose pela UDP-glicose-pirofosforilase. A
partir de uma unidade iniciadora (“primer”) e com o “auxilio” da glicogenina, forma-se, por
adição sucessiva de unidades de UDP-glicose, em que posteriormente o UDP é libertado, uma
cadeia linear, de ligações alfa-1,4. Posteriormente, por acção da enzima ramificante, formamse ligações alfa-1,6, dando a conformação final ramificada do glicogénio.
Glicogenólise: é a degradação do glicogénio de modo a obter moléculas de glicose, em caso
de necessidade. Por acção da glicogénio fosforilase, reduz a cadeia de glicogénio a menos
uma unidade, formando também uma molécula de Glicose-1-fosfato. Não é o inverso do
processo anterior, essencialmente porque temos neste caso um processo directo, sem o
intermediário UDP-Glicose.
As fosforilases hepáticas são diméricas – a e b, diferenciadas pelo facto de a primeira
ser fosforilada (fosfoserina) e de a segunda não (serina).
Nos lisosomas a cisão do glicogénio faz-se por hidrólise e não por fosforólise, por uma
maltase ácida.
O ciclo de Cori engloba os vários processos que ocorrem quer no músculo quer no
fígado. No músculo a glicose proveniente do sangue entra em processo de glicólise, com a
libertação de 2 ATP’s. As duas moléculas de ácido pirúvico formadas entram então num
processo de fermentação láctica, durante o qual o NADH é re-oxidado, regenerando o NAD+,
algo essencial para que a glicólise se continue a processar. O lactato produzido irá então
entrar na corrente sanguínea, estando disponível para a outra parte do ciclo de Cori - no
fígado, que engloba as reacções anteriormente referidas. Este ciclo é bastante importante
porque, apesar de ter um fraco rendimento energético, previne a acidose láctica no músculo
sob condições anaeróbias. Existe um ciclo análogo ao de Cori ainda menos produtivo que é o
ciclo da alanina. Neste ciclo, no músculo a partir do piruvato é produzida alanina por
transaminação, que depois através do sangue irá chegar ao fígado onde sofrerá a reacção
inversa.
Em termos de regulação, temos por exemplo a acção específica da glicagina no fígado (onde
também actua com o mesmo efeito a epinefrina), que, via AMPc, inibe a glicólise, e será
então importante em situações de necessidade energética, como veremos adiante.
Ainda na glicólise, sabe-se que a falta de ATP, ou predominância de AMP estimula o
processo, tal como o faz a frutose-2,6-bisfosfato, que como já vimos, inibirão a
gliconeogénese, estimulada por sua vez, pelo citrato e acetil-CoA. Para referir a regulação da
glicogénese/glicogenólise, a glicagina também desempenha um papel importante, uma vez
que activa a glicogenólise (para obter glicose), estimulando as PKA’s, que ao fosforilarem a
glicogénio fosforilase, activam-na, inibindo também a glicogénese ao fosforilarem
(inactivando) a glicogénio sintase. Numa via não dependente de AMPc, temos de libertar
cálcio (que promove a fosforilação, estimulando também a glicogenólise): hormonas como a
vasopressina e a epinefrina, ao se ligarem a receptores específicos da membrana, activam o
IP3 (fosfatidilinositol-4,5-trifosfato) e o 1,2-diacilglicerol, em que o IP3 promove a libertação
do cálcio do retículo endoplasmático e activa a fosforilase cinase, e o segundo activa a
proteína cinase.
No músculo, não existe glicagina apenas a epinefrina que irá estimular os receptores
adrenérgicos β favorecendo a transformação de ATP em AMPc. O AMPc irá activar as
PKA’s, estimulando a glicogenólise e inibindo da glicogénese.
Ainda no músculo o aumento de Ca2+ além de estimular a contracção muscular, estimula
também um dado tipo de cinases proteícas – “as cinases proteicas dependentes da
calmodulina” que irão ter o mesmo efeito: a estimulação da glicogenólise e inibição da
glicogénese.
Outra hormona muito importante, referida também adiante, é a insulina, que por um processo
inverso ao anterior, estimula a glicogénese e inibe a glicogenólise.
93
Regulação da Glicemia em estado alimentado
A glicemia varia consoante o estado fisiológico do organismo, dependendo de factores como
a alimentação ou o exercício físico, entre outros. Muitas formas de stress intenso e trauma,
AVC, ataque cardíaco e cirurgia podem aumentar temporariamente os níveis de glicose. A
glicemia pode também variar com o uso de certas drogas, aumentando com o uso de anti
depressivos, corticosteroides, diuréticos, estrogénios, lítio,etc ou diminuindo com o uso de
outras, como álcoois, esteróides, paracetamol,etc. refira-se por curiosidade que a doença mais
associada ao aumento crónico da glicemia é a “diabetes metillus”. Mas vamos nos focar na
regulação da glicemia após a alimentação, em que há um aumento temporário dos níveis de
glicose, atingindo uma concentração de 6.5 a 7.2 mmol/L.
O pâncreas é essencial na regulação do metabolismo energético do organismo, ao secretar e
lançar na corrente sanguínea as hormonas peptídicas essenciais à mesma, sendo a insulina e a
glucagina as principais. A insulina é uma hormona polipeptídica fabricada nas células β dos
ilhéus de Langerhans no pâncreas que regula o metabolismo dos hidratos de carbono. É o
principal agente na homeostase dos hidratos de carbono. A quantidade de insulina em
circulação tem efeitos extremos espalhados por todo o corpo. Concentrações elevadas de
glicose como as que ocorrem logo após a alimentação estimulam a secreção de insulina. Deste
modo a percebe-se que a regulação da glicemia neste estado e feita principalmente através da
insulina.
A regulação da síntese de insulina é efectuada através dos seguintes mecanismos: a glicose
entra nas células beta pelo transportador de glicose GLUT2, depois passa pela glicólise e pelo
ciclo respiratório, onde são produzidas moléculas de ATP. Então os canais de potássio como
são controlados pelo ATP vão-se fechar e a membrana celular vai ser despolarizada. Sob
despolarização, os canais de cálcio (Ca2+) controlados pela voltagem eléctrica abrem-se e os
iões de cálcio difudem-se para dentro das células. O aumento do nível de cálcio causa a
activação da fosfolipase C, que corta o fosfolipídeo da membrana fosfatidil inositol 4,5bifosfato em 1,4,5-trifosfato e diacilglicerol. O inositol 4,5-bifosfato liga-se às proteínas
receptoras no retículo endoplasmático. Isto aumenta ainda mais a concentração de cálcio no
interior da célula. O aumento significativo de cálcio na célula produz a libertação de insulina
previamente sintetizada, que tinha sido armazenada em vesículas secretoras. O nível de cálcio
também controla a expressão do gene de insulina via proteína Ligante de Elemento
Responsivo a Cálcio (em inglês, CREB)
Falta então explicar de que forma e que a insulina promove a diminuição da glicemia: A
insulina liga-se ao seu receptor na membrana do hepatócito e do músculo desencadeando
cascatas de activações enzímicas que desembocam na activação da fosfatase-1 de proteínas e
na inactivação da cínase-3 da síntase do glicogénio. A activação da fosfatase-1 de proteínas
vai provocar um conjunto de desfosforilações (na síntase do glicogénio, na cínase da
fosforílase e na fosforílase do glicogénio) que promovem a glicogénese e travam a
glicogenólise, fazendo com que a glicose seja temporariamente armazenada sob a forma de
glicogénio. A cínase-3 da síntase do glicogénio é uma das enzimas envolvidas na inactivação
da síntase de glicogénio: a inactivação desta cínase também contribui para a promoção da
glicogénese. A insulina suprime também a produção de glucagina e, consequentemente, reduz
a produção hepática de glicose, sendo assim bastante eficaz na redução da glicemia.
A regulação da secreção de insulina é um processo delicado que é condicionado por muitas
variáveis. De algumas já falámos, por exemplo a subida da glicemia estimula a produção e a
descida inibe. Adicionalmente, parte da síntese e libertação de insulina pode ocorrer na
ausência de glicose ou carbohidratos , e as células beta são também ainda influenciadas de
alguma forma pelo sistema nervoso autónomo. A acetilcolina, ejectada das terminações
nervosas do sistema nervoso parassimpático, a colecistocinina, difundida por células
94
enteroendócrinas da mucosa intestinal, e o peptídeo inibitório gastrointestinal são algumas
substâncias que estimulam também a libertação de insulina. O sistema nervoso simpático
(agonistas adrenérgicos alfa-2) pode por seu turno inibir a libertação de insulina. A libertação
de insulina é fortemente inibida pela hormona relacionada com o stress, a epinefrina, e o
exercício físico ao consumir bastante glicose também inibe a produção de insulina. Por fim,
quando a glicemia se estabelece nos valores fisiológicos normais, cessa ou diminui a
produção de insulina a partir das células beta. Se a glicemia cai abaixo desses valores
normais, especialmente a valores perigosamente baixos, a libertação de hormonas
hiperglicemiantes vai induzir à disponibilização de glicose ao sangue, como vamos ver a
seguir.
Regulação da Glicemia em jejum
O cenário oposto ao estado alimentado corresponde ao estado de jejum; neste, vai
ocorrer uma queda dos níveis de glicémia, para valores entre os 3.3 e os 3.9mmol/L, o que
desencadeia um estado de hipoglicémia. Este estado de hipoglicémia, se prolongado, vai levar
à utilização e possível esgotamento das reservas energéticas do fígado, o que tem
consequências muito graves a nível cerebral, podendo as disfunções cerebrais iniciais evoluir
para um estado de coma, que pode levar à morte.
A regulação dos níveis de glicémia na situação de jejum, é processada
maioritariamente a nível hormonal, destacando-se a glicagina como hormona principal desta
via de regulação da glicémia. A glicagina é produzida na porção endócrina do pâncreas,
nomeadamente nas células α dos ilhéus de Langerhans, apresentando acções antagonistas às
da insulina, o que faz sentido pelo facto de serem libertadas como resposta a níveis opostos de
glicémia. O esquema seguinte ilustra de uma forma geral os mecanismos de acção da
glicagina perante níveis baixos de glicémia, como em situação de jejum:
Acção da glicagina
Seguindo o esquema, observamos que perante uma descida dos níveis de glicémia, vai
haver libertação de glicagina que vai, por via da adenilato ciclase, gerar AMPc que activa a
proteina cinase 1(PKA). Esta vai ser mediadora de todos os efeitos da glicagina, através de
vários processsos de fosforilação. A inibição da glicólise é conseguida por duas vias
diferentes; pela fosforilação e inactivação directa da enzima glicolítica piruvato cinase e pela
inactivação da PFK-1 através da fosforilação da proteína bifuncional PFK-2/FBPase-2 e
consequente activação da FBPase-2, que diminui a concentração de Frutose 2,6- bifosfato.
Estas acções da glicagina levam, não só à inibição da glicólise, como também, à estimulação
da gliconeogénese. Por sua vez, a estimulação da glicogenólise é conseguida através da
activação da fosforilase b cinase, que vai activar a forma activa da fosforilase (forma a) por
fosforilação de uma forma menos activa (forma b), o que vai levar à formação de glicose 6fosfato. No fígado, este composto pode ser transformado em glicose devido à presença de
95
glicose 6-fosfatase, e posteriormente libertado para a corrente sanguínea com o intuito de
aumentar os níveis de glicémia. De referir ainda, o facto, da fosforilase a ter dois locais
alostéricos de ligação a glicose, o que a leva funcionar como um sensor de glicose no fígado.
Por fim, a inactivação da glicogénese, é conseguida através da diminuição da
desfosforilação da glicogénio sintetase b (inactiva) e pela fosforilação da glicogénio sintetase
a (activa). Estas acções privilegiam a forma b da glicogénio sintetase, inactivando, desta
forma o processo de glicogénese.
96
3.12- DERIVADOS LIPÍDICOS ESPECIAIS
(a) Sinais metabólicos de natureza lipídica-estrutura,síntese e actividade
Existem diversas classes de lípidos no nosso organismo. Os lípidos de reserva e
estruturais são importantes constituintes celulares: lípidos de membrana constituem cerca de
5%-10% da massa total de grande parte das células, enquanto que os lípidos de reserva
chegam a constituir mais de 80% da massa total do adipócito. Excluindo algumas importantes
excepções, estas classes lipídicas desempenham um papel passivo na célula. Outro grupo de
lípidos, presentes em muito menores quantidades, tem um papel activo, funcionando como
potentes sinais intra e extracelulares.
Um grupo muito importante de sinais intracelulares de natureza lipídica são os
glicerofosfolipidos (nomeadamente o fosfatidilinositol), cuja estrutura geral é a seguinte:
através de ligações éster, encontram-se ligados ao carbono 1 e 2 do glicerol um ácido gordo
saturado e um insaturado, respectivamente. Ao carbono 3 encontra-se ligado um grupo
substituinte, através de uma ligação fosfodiéster (no caso do fosfatidilinositol, o grupo
substituinte é o inositol).
O fosfatidilinositol encontra-se na membrana plasmática sob a forma de
fosfatidilinositol 4,5- bisfosfato, que actua em resposta a um sinal hormonal que chega à
membrana celular e liga-se a um receptor específico, causando a conversão do GDP em GTP
da proteína Gq , causando a dissociação desta do receptor. Ela vai activar a fosfolipase C que
actua sobre o fosfatidilinositol 4,5- bisfosfato, clivando a ligação fosfodiéster entre o inositol
4,5 – bisfosfato e o glicerol, originando dois mensageiros secundários: diacilglicerol e inositol
1,4,5-trifosfato (IP3). Este último difunde para o citosol, ligando-se a receptores específicos
do retículo endoplasmático, abrindo canais de Ca2+, aumentando a concentração deste ião no
citoplasma. Entretanto, devido à sua natureza lipídica, o diacilglicerol movimenta-se
livremente na zona hidrofobica da membrana celular, onde se vai ligar à proteína cinase C, e
em conjunto com o aumento da concentração de Ca2+ vai activa-la. Caso o cálcio libertado
pelo retículo endoplasmático não seja suficiente, o IP3 desencadeia a abertura dos canais de
Ca2+ da membrana celular. Por fim a proteína cinase C vai fosforilar resíduos Ser e Thr de
proteínas alvo, desencadeando assim a resposta celular. Este mecanismo chama-se cascata dos
fosfoinositois.
Outro grupo de sinais metabólicos de natureza lipídica são os eicosanoides, que
funcionam como hormonas parácrinas e autócrinas, ou seja, actuam ou na célula que os
produz ou nas células adjacentes, em vez de serem libertados para a circulação. Estes
derivados de ácidos gordos têm uma grande variedade de efeitos dramáticos nos tecidos: estão
envolvidos no processo de reprodução; na inflamação, febre e dor associados a doença ou
lesão; na coagulação e regulação da pressão sanguínea; na secreção ácida entre outras.
São todos derivados do ácido araquidónico, libertado por fosfolípidos de membrana,
por acção da fosfolipase A2, em resposta a sinais hormonais. São compostos de 20 carbonos e
existem 3 classes: Prostaglandinas, Tromboxanos e Leucotrienos. A formação dos dois
primeiros é inibida por drogas anti-inflamatórias não-esteoides, por inibição da enzima
ciclooxigenase, que cataliza um dos primeiros passos da formação destes dois compostos.
As Prostaglandinas contêm um anel de 5 carbonos, originado pela ligação dos carbonos 8 e 12
do ácido araquidónico. Existem dois grupos de Prostaglandinas: PGE (solúvel em éter) e PGF
(solúvel em tampão fosfato), cada um destes contendo numerosos subtipos. Actuam em
numerosos tecidos por regulação da síntese de cAMP, e dado que este composto é o mediador
da acção de diversas hormonas, as Prostaglandinas afectam uma grande variedade de funções
celulares tais como a contracção do músculo liso do útero, elevação da temperatura corporal
(febre) e causam inflamação e dor.
Os tromboxanos têm um anel de 6 membros, contendo um éter, formado pela ligação dos
carbonos 8 e 12 do ácido araquidónico. São produzidos pelas plaquetas e actuam na formação
de coágulos sanguíneos e na redução do fluxo de sangue para o local de coagulação.
97
Os Leucotrienos têm uma série de ligações duplas conjugadas. Foram inicialmente
descobertas nos leucócitos e são potentes sinais biológicos, com diversas acções,
nomeadamente a contracção dos músculos que envolvem as vias respiratórias, podendo causar
ataques asmáticos, por excesso de produção.
Os esfingolípidos de membrana também funcionam como fonte de sinais intracelulares. A
estrutura de um esfingolípido consiste na ligação de um ácido gordo ao carbono 2 da
esfingosina, e ao carbono 3 encontra-se ligado um grupo substituinte, por uma ligação éster.
Os esfingolípidos originam-se através de reacções da ceramida (o grupo substituinte é um
átomo de H). Nomeadamente, a esfingomielina (constituinte das bainhas de mielina que
revestem o axónio), forma-se através da reacção da ceramida com a fosfatidilcolina,
originando também diacilglicerol, um importante segundo mensageiro como já foi falado (o
grupo substituinte da esfingomielina é a fosfocolina). A ceramida e a esfingomielina são
reguladores potentes de proteínas cinases, e a primeira está envolvida na regulação da divisão,
diferenciação e migração celular, e também na apoptose.
Por fim falando de hormonas esteróides. São produzidas essencialmente nas gónadas e suprarenal, sendo libertadas para a circulação sanguínea, onde se ligam a transportadores proteicos,
sendo assim transportadas para os tecidos alvo. São derivados do colesterol, mais
concretamente da pregnolona. Dada a sua natureza lipídica (as hormonas esteroides mantêm o
núcleo esterol do colesterol) difundem facilmente através da bicamada fosfolipídica, ligandose a receptores específicos do núcleo, desencadeando mudanças na expressão genética e
metabolismo. Dada a sua elevada especificidade para esses receptores, apenas quantidades
mínimas destas hormonas são produzidas.
b) Conversão enzimática do colesterol em precursores
e hormonas esteroides-estimulação, etapas, localização celular e
caracterização dos produtos
A síntese de hormonas esteróides tem como precursor principal o
colesterol, composto por vinte e sete átomos de carbono
numerados segundo a ordem apresentada no diapositivo 13, esta
ordem é bastante importante para perceber os mecanismos de
formação deste tipo de hormonas.
Os locais de síntese destas hormonas são principalmente:
glândulas supra-renais, gónodas, fígado, pele e rins.
A nível celular os processos são idênticos nos vários locais de síntese, o colesterol
para esse processo provém em grande parte da porção deste que se encontra em circulação e
da conversão do piruvato através de um processo complexo que envolve o melonovato. A
estimulação do processo de síntese de hormonas esteróides é da responsabilidade de
hormonas proteicas hipofisárias, LH (Luteotropic hormone) no caso das gónodas e ACTH
(adrenocorticotropic hormone) no caso das glândulas supra-renais e a angiotensina II, um
péptido derivado da angiotensina que estimula especialmente a via dos mineralocorticóides. A
síntese desta classe de hormonas não se dá apenas num organelo celular, normalmente estão
envolvidos mitocôndria e retículo endoplasmático, como tal o colesterol (molécula
hidrofóbica) tem que atravessar regiões em solução aquosa (espaço inter-membranar
mitocondrial), para tal um complexo proteico denominado de StAR (steroidogenic acute
regulatory, cuja síntese é influenciada positivamente pelos factores estimulantes) liga-se ao
colesterol tornando assim possível a sua difusão através desses meios desfavoráveis.
O mais complexo e principal órgão de produção são as glândulas supra-renais. Nestas
estruturas mais precisamente no seu cortéx são sintetizados vários tipos de hormonas
esteróides: mineralocorticóides (mais abundantemente na zona glomerular do cortéx)
glicocorticóides e androgénios (principalmente nas zonas reticular e fasciculada), pode
98
também haver formação de estrogénios sendo esta a principal fonte deste tipo de hormonas
após a menopausa.
O primeiro passo na síntese de hormonas esteróis é a conversão de
colesterol em pregnenolona, esta reacção mediada pelo complexo
enzimático citocromo P450scc (side chain clivage) na membrana interna
da mitocôndria, esta reacção envolve duas hidroxilações e uma clivagem
da sequência linear do colesterol, formando a pregnenolona uma molécula
de 21 carbonos percursora de todas as hormonas esteróides.
Como já referimos existe uma especialização das regiões do cortéx
supra-renal, na zona glomerular dá-se a síntese de mineralocorticóides, que
tem reacções em comum com os glicocorticóides que por sua vez têm com
os androgénios. A verdade é que não podemos distinguir claramente três
vias, mas sim um sistema complexo em que mesmo subprodutos têm
actividade, sendo os produtos finais apenas compostos mais activos
resultantes de reagentes menos activos, como é o caso da corticosterona
que tem mais carácter de glicocorticóide que de mineralocorticóide, mas que é o percursor da
aldosterona. Tomando mesmo este exemplo, podemos ver que através do complexo
enzimático 3β – (OHSD) e a Δ5,4 isomerase a pregnenolona é transformada em progesterona
que depois hidroxilações nos carbonos 11, 21, não sendo possível seguir a via dos
glicocorticóides porque a zona glomerular não possui o complexo enzimático citocromo P450
C17 capaz de hidroxilar o carbono 17, responsável pelo
carácter de glicocorticóide da molécula. Porém esta
Via dos mineralocorticóides –
Via dos glicocorticóides –
Via dos androgénios –
zona possui um complexo mitocondrial, aldosterona sintase capaz de hidroxilar o carbono 18
convertendo assim a corticosterona num mineralocorticóide mais poderoso. Quanto à zona
fasciculada e à zona reticular sabemos que possuem uma estrutura enzimática conhecida
como citocromo P417 C17, constituído por uma liase e hidroxilase que actuam no carbono 17
da molécula, estas enzimas vão ser cruciais tanto na via dos androgénios como na via dos
glicocorticóides, formando cortisol o glicocorticóide mais potente na espécie humana e o
principal percursor de androgénios supra-renal a desidroepiandrosterona. Normalmente a via
dos glicocorticóides predomina em relação à dos androgénios, mas problemas a nível de umas
das enzimas da segunda podem provocar excesso de produção de androgénios, os androgénios
supra-renais são normalmente transformados na mais potente testosterona através da redução
do seu percursor, androstenediona, no carbono 17, apenas pequenas quantidade são
produzidas nos nas supra-renais sendo a maioria nos testículos.
A nível testicular a síntese dá-se nas células de Leydig, o mecanismo à bastante
parecido com que ocorre nas supra-renais, sendo o estimulador neste caso a LH, a nível
99
testicular a formação de testosterona pode seguir duas vias, a Via da Progesterona ou Δ4
via da desidroepiandrosterona ou Δ5, sendo a segunda preferencial. Como produto final da
testosterona temos que do seu metabolismo pode resultar a Dihidrotestosterona (DHT), ou
17-cetoesteróis, os segundos acontecem no fígado e são compostos com muito menos
poder que o seu percursor, no entanto o primeiro é muito mais potente que a testosterona
sendo aliás a sua forma activa em locais como a próstata, órgãos genitais e certas zonas da
pele. A reacção é mediada pela 5-α reductase com NADH envolvido.
Os estrogénios são uma família de hormonas derivadas dos androgénios por
aromatização, podem ser produzidos nas glândulas supra-renais, células adiposas, fígado,
placenta e nas mulheres em idade fértil nos folículos. A origem dos estrogénios periféricos
(fora das gónodas) é aromatização de testosterona e seus percursores, sendo de principal
importância em indivíduos que não possuem por alguma razão esteroido-génese folicular.
Em termos foliculares a proveniência dos estrogénios resulta de uma interacção entre as
células da teca e da camada granulosa na qual as primeiras produzem androgénios, em
especial testosterona que as segundas sintetizam em estrogénios em especial o 17βestradiol.
100