Imagem da Semana: Fotografia e radiografia
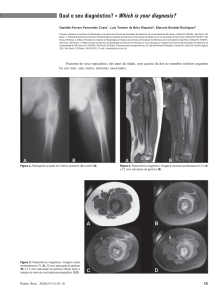
Figura 1: Fotografia de hemiface esquerda
Figura 2: Fotografia de couro cabeludo
Figura 3: Radiografia de crânio em perfil esquerdo
Enunciado
Paciente feminina, 2 anos, apresenta nódulos em couro cabeludo e em região
infraorbitária esquerda de crescimento progressivo e doloroso há sete meses,
associado à dermatite seborreica em couro cabeludo. Há quatro meses evoluiu
com quadro de polidipsia, poliúria e otorreia crônica bilateral, não responsiva a
antibioticoterapia. Previamente hígida. Foi realizado estudo radiológico do crânio.
Com base na história clínica e nas imagens apresentadas, o diagnóstico
mais provável é:
a) Histiocitose de células de Langerhans
b) Xantogranuloma juvenil
c) Síndrome hemofagocítica
d) Neuroblastoma
Análise da imagem
Imagem 4: Fotografia de hemiface esquerda evidencia nódulo de aproximadamente
2,0cm de diâmetro, de coloração violácea e base infiltrada (seta vermelha), e pápula de
aproximadamente 0,5cm de diâmetro e limites bem definidos (seta preta). As lesões são
dolorosas, sem outros sinais flogísticos.
Imagem 5: Fotografia de couro cabeludo revela lesões papuloescamosas, eritematosas e
amareladas, cobertas por escamas gordurosas, sugestivas de dermatite seborreica.
Imagem 6: Radiografia de crânio em perfil esquerdo evidenciando lesões osteolíticas
múltiplas de margem irregular em calota craniana.
Diagnóstico
A manifestação clínica mais comum da histiocitose de células de Langerhans
(HCL) é o acometimento ósseo. As lesões são osteolíticas e o crânio é o principal local
acometido, o que justificou a realização da radiografia de crânio na paciente. O diabetes
insipidus é a endocrinopatia mais prevalente na HCL e outra manifestação neurológica
comum é a otite média recorrente. O envolvimento cutâneo é observado em mais de um
terço das crianças e as regiões mais afetadas são o couro cabeludo e as zonas
intertriginosas. A morfologia clínica das lesões é muito variável e a dermatite seborreica
de difícil tratamento também é um sinal comum da doença.
O xantogranuloma juvenil é a doença mais frequentemente confundida com a HCL e
manifesta-se como uma lesão cutânea papulosa firme, única, com telangiectasia
superficial. A ocorrência de múltiplas lesões e o envolvimento sistêmico grave são pouco
comuns.
As síndromes hemofagocíticas são desordens relacionadas à ativação inapropriada
de macrófagos. Os critérios para o diagnóstico das síndromes hemofagocíticas são: febre
sem outra causa definida, esplenomegalia,
citopenias, hipofibrinogenemia ou
hipertrigliceridemia e hemofagocitose demonstrada na medula óssea, baço, linfonodo ou
outro tecido.
O neuroblastoma é o tumor sólido extracraniano mais frequente na infância. Originase a partir de células da crista neural e pode surgir em qualquer ponto do neuro-eixo. São
manifestações comuns em doença disseminada: irritabilidade, anorexia, perda de peso,
palidez, dores ósseas e febre. Hipertensão, geralmente não acentuada, é vista em até 50%
dos casos.
Discussão do caso
A histiocitose de células de Langerhans (HCL) é uma doença hematológica rara e resulta
da proliferação clonal das células de Langerhans. Sua etiopatogenia é desconhecida e há
hipóteses de que seja uma doença neoplásica ou de origem inflamatória, com possível
existência de fatores predisponentes imunológicos, virais ou genéticos. Pode ser
diagnosticada em qualquer faixa etária, acometendo principalmente crianças de um a
três anos. A incidência anual na população pediátrica é estimada em três a quatro casos
por milhão.
A apresentação clínica e a evolução da HCL são muito variáveis. Praticamente todos os
tecidos podem ser acometidos pela doença, que pode se manifestar como lesão isolada
em um único órgão ou como doença disseminada com disfunção orgânica. Os órgãos
mais acometidos ao diagnóstico são: ossos (77%), pele (39%), linfonodos (19%), fígado
(16%), baço (13%), mucosa oral (13%), pulmões (10%), sistema nervoso central (6%). A
forma multissistêmica afeta principalmente crianças menores de três anos. A doença
pode regredir espontaneamente ou evoluir com comprometimento da função de órgãos
vitais, com consequências graves e fatais.
O tempo desde o início das manifestações clínicas até o diagnóstico pode ser longo,
levando de 5 a 20 anos. Frequentemente, o diabetes insipidus é o sintoma inicial e não é
atribuído à HCL até que outros sintomas apareçam. O diagnóstico é feito por meio de
história clínica compatível associada à biópsia das lesões.
Conforme a apresentação clínica, o tratamento pode variar desde conduta expectante,
excisão cirúrgica das lesões, corticoterapia, quimioterapia, combinação de todos os
anteriores e até transplante de medula óssea. O tratamento quimioterápico, quando
indicado, pode reduzir o número de recidivas e sequelas nos casos de doença
multissistêmica, doença óssea multifocal e doença localizada nos denominados sítios
especiais (ossos faciais e da fossa craniana média e lesões com extensão intra-espinhal).
Nos casos de doença em um único sistema, o tratamento é individualizado.
Aspectos relevantes
- A histiocitose de células de Langerhans (HCL) é uma doença hematológica rara e
acomete principalmente crianças de um a três anos de idade;
- Os órgãos mais acometidos são ossos, pele e linfonodos;
- As lesões ósseas típicas são compostas de histiócitos gigantes multinucleados do tipo
osteoclasto, com destruição óssea, necrose, hemorragia e abscessos eosinofílicos;
- O diagnóstico é feito pela história clínica associada à confirmação histológica das
lesões;
- O tratamento é variável e engloba desde a conduta expectante até quimioterapia e
transplante de medula óssea.
- Apesar de rara, a hipótese de HCL deve ser aventada especialmente diante de casos
de diabetes insipidus, de dermatite seborreica e de otorreia crônica refratárias ao
tratamento.
Referências
- Campos MK, Viana MB, Oliveira BM, Ribeiro DD, Silva CMR. Langerhans cell
histiocytosis: a 16-year experience. Jornal de Pediatria. 2007;83(1):79-86.
- Babeto LT. Histiocitose das Células de Langerhans na criança: revisão dos achados
clínicos, anátomo-patológicos e imuno-histoquímicos dos casos diagnosticados no
Hospital das Clínicas da UFMG – 1988 a 2008. Belo Horizonte: 2009.
- McClain KL. Clinical manifestations, pathologic features, and diagnosis of Langerhans
cell histiocytosis. Boxer LA, Park JR, editores. Walthman (MA): UpToDate; 2012.
Disponível em: http://www.uptodate.com/contents/clinical-manifestations-pathologicfeatures-and-diagnosis-of-langerhans-cell-histiocytosis
- McClain KL. Treatment of Langerhans cell histiocytosis.Boxer LA, Park JR, editores.
Walthman
(MA):
UpToDate;
2012.
Disponível
http://www.uptodate.com/contents/treatment-of-langerhans-cell-histiocytosis
em:
Responsáveis
Camila Gomes de Souza Andrade, acadêmica 12º período de Medicina da FM-UFMG
E-mail: camila-gomes[arroba]ufmg.br
Dra. Bruna Faria Vieira, médica, graduada pela FM-UFMG
E-mail: brunafariavieira[arroba]gmail.com
Dra. Ana Carolina Bueno e Silva, residente de Pediatria do HC-UFMG
E-mail: acbuenos[arroba]yahoo.com.br
Orientadoras
Profa. Karla Emília de Sá Rodrigues, pediatra, professora do Departamento de Pediatria
da FM-UFMG
E-mail: karla.emilia[arroba]uol.com.br
Profa. Benigna Maria de Oliveira, pediatria, professora do Departamento de Pediatria da
FM-UFMG
E-mail: benigna[arroba]uol.com.br
Revisores
Fabiana Resende, Emília Valle, Profa. Viviane Parisotto