VINÍCIUS BURNETT – MED 92 HEMATOLOGIA CÉLULA-­‐TRONCO HEMATOPOIÉTICA E HETEROGENEIDADE DAS CÉLULAS-­‐TRONCO 19/08/2011 MEDULA ÓSSEA SÉRIE MIELOIDE •
•
•
•
•
Megacariócito; Neutrófilo; Eosinófilo; Basófilo; Monócito. SÉRIE LINFOIDE •
•
•
Linfócito B; Linfócito T; Células NK. CÉLULA-­‐TRONCO HEMATOPOIÉTICA (CTH) CARACTERÍSTICAS •
•
•
Autorrenovação: atividade da telomerase; Diferenciação multilinhagem: célula-­‐tronco hematopoiética pode originar os progenitores das séries linfoide e mieloide, que darão origem às células sanguíneas. Reconstituição de tecido em um sistema irradiado: podemos destruir todas as células do sangue e da medula óssea (no tratamento de leucemias, por exemplo). Depois, ao se fazer um transplante de medula, as células implantadas são capazes de “colonizar” os ossos do indivíduo e gerar células sanguíneas novamente. IDENTIFICAÇÃO •
•
•
A célula-­‐tronco hematopoiética possui os seguintes marcadores de superfície: CD34 +, CD38 -­‐, CD138 +, Lin -­‐. Possui a capacidade de efluir o corante rodamina ! Garante proteção à célula; " O problema é que a CTH também é capaz de expulsar quimioterápicos, o que dificulta o tratamento com essas drogas em casos de cânceres. Manutenção em certos meios de cultura. VINÍCIUS BURNETT – MED 92 DESTINO DA CÉLULA-­‐TRONCO HEMATOPOIÉTICA •
•
•
Três possibilidades: " Apoptose; " Diferenciação; " Autorrenovação, que pode ser simétrica (quando a CTH origina duas outras CTHs idênticas) ou assimétrica (quando a CHT origina uma CTH e uma outra célula, que pode se diferenciar ou entrar em apoptose). Como saber o caminho que a célula “escolhe”? " Teoria estocástica: diz que a escolha é aleatória; " Teoria indutora: diz que o meio induz a célula a escolher determinado caminho. Ao se dividirem, as células-­‐tronco dão origem às células precursoras, as quais se caracterizam pela perda do potencial de autorregeneração e pelo comprometimento com uma dada via de diferenciação. INFLUÊNCIAS SOBRE A CTH – FATORES DE CRESCIMENTO •
•
•
•
Fatores de crescimento são glicoproteínas secretadas pelas células do estroma e que atuam na sobrevivência, proliferação e diferenciação das células hematopoiéticas; Os fatores de crescimento são divididos em duas grandes famílias, conforme o tipo de receptor de membrana sobre os quais atuam: família das citosinas e família tirosina quinase; Entre os fatores de crescimento mais relevantes para a proliferação estão: ligante Kit, GM-­‐CSF, IL-­‐3, IL-­‐4, IL-­‐6. A ação desses fatores resulta no recrutamento de células-­‐tronco e na proliferação e diferenciação em precursores intermediários (multilinhagem); A diferenciação em precursores unilinhagem requer fatores específicos: " Mielopoese: G-­‐CSF e M-­‐CSF; " Eritropoese: Eritropoetina (Epo); " Linfopoese: • IL-­‐6 e IL-­‐7 ! linfócitos B; • IL-­‐2 e IL-­‐3 ! linfócitos T. VINÍCIUS BURNETT – MED 92 GRANULOCITOPOESE 24/08/2011 CÉLULAS GRANULÓCITAS •
•
Neutrófilo, basófilo e eosinófilo; Todos possuem núcleos polilobulados. NEUTRÓFILO CARACTERÍSTICAS •
•
•
•
•
•
•
•
Tamanho um pouco maior que o das hemácias; Possui três lobulações; Constitui o tipo mais comum de leucócito; É produzido, “maturado” e armazenado na medula óssea; Possui meia-­‐vida de 7 horas; A reserva medular é de 10 a 15 vezes maior do que a quantidade encontrada na corrente sanguínea; A reserva periférica, cujo principal componente é o baço, é de 2 vezes a quantidade encontrada em circulação; Mecanismo de ação: reconhece algo estranho ! fagocita ! degranula ! gera uma resposta inflamatória local. PROLIFERAÇÃO E DIFERENCIAÇÃO PELA REGULAÇÃO POR CITOCINAS •
•
•
As principais citocinas que medeiam a maturação de neutrófilos são a G-­‐CSF (fator estimulador de colônias de granulócitos) e o GM-­‐CSF (fator estimulador de colônias de granulócitos e macrófagos). Esses receptores são ativados por meio do sistema JAK-­‐STAT; Estudo em ratinho sem G-­‐CSF: o animal (praticamente) deixa de gerar neutrófilos; Estudo em rato sem GM-­‐CSF: o animal continua a gerar granulócitos. " Importância do GM-­‐CSF: em pacientes fazendo quimioterapia, a aplicação de GM-­‐CSF ajuda na recuperação. VINÍCIUS BURNETT – MED 92 FATORES TRANSCRICIONAIS •
•
•
São glicoproteínas capazes de reconhecer a região promotora do DNA e mediar a produção de proteínas específicas. Fatores gerais: " SCL, GATA-­‐2, AML-­‐1. • AML-­‐1: " Junto com o C-­‐EBPα, é um dos fatores mais importantes para a formação dos granulócitos; " Liga-­‐se ao DNA, dimeriza-­‐se e induz a transcrição. Fatores comprometidos com uma série específica: " GATA-­‐1, PU.1, C-­‐EBPα (CCAAT-­‐Enhancer Binding Protein α) • C-­‐EBPα: " Liga-­‐se a sequências 5’TKN NGYAAK3’ e é o principal regulador da granulocitopoese. PROCESSO MATURATIVO DOS NEUTRÓFILOS VINÍCIUS BURNETT – MED 92 IDENTIFICAÇÃO IMUNOFENOTÍPICA •
Consiste na identificação de marcadores de superfície que indicam imaturidade, linhagem e maturidade. "
"
"
Marcadores de imaturidade: • CD 34, CD 117, HLA-­‐DR Marcadores de linhagem: • CD 13, CD 33 Marcadores de maturidade: • CD 11b, CD 16 • Aparecem no fim do processo de maturação e são específicos de cada linhagem. Observação: pensar na seguinte célula: •
•
•
CD 34+, CD 13+, CD 33+, CD 15, CD 11b+, CD 16. Como se vê, a célula possui características de adulta, mas apresenta o marcador de imaturidade CD 34. A explicação é que se trata de uma célula neoplásica; Esse marcador é útil para se verificar a doença residual mínima, que consiste no número de células neoplásicas encontrado após o tratamento do câncer. TIPOS DE GRÂNULOS •
Grânulos azurófilos (primários): " Mucopolissacarídeos sulfatados; " São os primeiros a aparecer; " São “grosseiros e basófilos”; " São corados intensamente pelo Azuro-­‐A; " Contém grande variedade de proteínas, como a mieloperoxidase, defensinas (arginina e cisteína), elastase. VINÍCIUS BURNETT – MED 92 •
•
•
Grânulos específicos (secundários): " Próprios de cada tipo de granulócito; " Lactoferrina; " Receptor de vitamina B12; " Proteínas inflamatórias: TNF, laminina, fibronectina, C3b; " Colagenase. Grânulos terciários: " Formados em estágios maduros; " Contém gelatinase, que destrói o tecido conjuntivo aonde o neutrófilo ativado se instalou. Dessa forma, aumenta a permeabilidade do tecido e destrói microrganismos. Vesículas secretórias: " Contém os seguintes receptores de membrana: • CD 14, CD 16, CD 11b, receptor de formilpeptídeo. " Contém proteínas plasmáticas; " Facilitam a chegada de células ao local infectado; " Não degranulam como os outros. PROCESSO INFLAMATÓRIO VINÍCIUS BURNETT – MED 92 EOSINÓFILO CARACTERÍSTICAS •
•
•
•
•
•
•
Correspondem a 3% dos leucócitos do sangue e da medula óssea; Contém duas lobulações; Grânulos muito mais grosseiros e alaranjados; Grânulos ricos em peroxidase, arilsulfatase, fosfatase ácida, fosfolipase e MBP; Processo de maturação igual ao dos neutrófilos; Responsável por mediar processos alérgicos; Responsável por mediar a destruição de parasitas; BASÓFILO CARACTERÍSTICAS •
•
•
•
•
Grânulos arroxeados e escassos; Menores que os neutrófilos; Promovem reações alérgicas imediatas; Histamina, serotonina, sulfato de controitina e leucotrienos; Medula óssea – sangue – tecidos. Observação: mastócito não vem do basófilo! PRIMEIRO SEMINÁRIO – SÍNDROME DE KOSTMANN (NEUTROPENIA CONGÊNITA GRAVE) •
•
•
Neutropenia: redução do número de neutrófilos (ANC < 500, quando o normal é entre 2000 e 7000); Fisiopatologia: defeitos em vários genes: " Gene ELANE: quando mutado, produz a elastase leucocitária, que desativa as proteínas “Notch” da via de diferenciação granulocítica. Isso causa uma paralisação no estágio promielócito; " Gene HAX-­‐1: produz uma proteína que evita a apoptose. Quando está mutado, a célula entra em processo de autodestruição. Sintomas: febre, tosse, ardor, ulcerações, calafrios, dor de garganta, irritabilidade, diarreia. VINÍCIUS BURNETT – MED 92 ERITROPOESE 26/08/2011 •
•
•
Consiste na maturação e diferenciação das hemácias; Começa igual à granulocitopoese; Primeiro sítio da hematopoese é o saco vitelínico: " Nos primeiros dois meses e meio de gestação, o saco vitelínico é o único responsável pela produção dos eritrócitos. Depois desse período, o fígado e o baço também passam a produzi-­‐los. Somente a partir do 5º mês de gestação, a medula óssea “assume o controle”; " Com o passar do tempo (a partir dos 30 anos de idade, aproximadamente), a eritropoese fica a cargo apenas dos ossos chatos: PROCESSO MATURATIVO VINÍCIUS BURNETT – MED 92 •
•
•
•
•
•
•
À medida que avança o processo de maturação, a cromatina se condensa e depois se perde o núcleo e o ácido nucleico. Normoblasto (ou proeritroblasto): " Citoplasma com muito RNA; " Núcleo grande; " CD 117 – imaturidade; " CD 45 – linhagem hematológica; " CD 71 – receptor de transferrina. Eritroblasto basófilo: " Maior condensação da cromatina; " Mais RNA. Eritroblasto policromático: " Cromatina continua a se condensar; " Último estágio em que há divisão celular. Eritroblasto ortocromático: " Citoplasma adquire a mesma coloração que a hemácia; " Núcleo é expulso num processo aparentemente ativo de extrusão. Reticulócito: " Cora organelas que não estão presentes nas células maduras; " Sem núcleo e funcional. Eritrócito: " CD 235a – marcador de maturidade. CÉLULAS ERITROIDES NA MEDULA ÓSSEA •
•
Os precursores de linhagem eritroide correspondem a cerca de 1/3 das células nucleadas da medula óssea; São produzidos cerca de 200 bilhões de reticulócitos por dia. PRECURSORES ERITROIDES •
•
•
Possuem alta capacidade proliferativa e intensa síntese proteica. Receptor de eritropoetina: " Estimula a proliferação de hemácias; " Expressão máxima nos proeritroblastos e nos eritroblastos basófilos. " BFU-­‐E: células precursoras nos estágios intermediários de diferenciação eritroide; " CFU-­‐E: células precursoras nos estágios finais de diferenciação eritroide. Receptor de transferrina: " Essencial para a incorporação de ferro pela célula. " Estão presentes onde há síntese proteica: desde os proeritroblastos até os reticulócitos. VINÍCIUS BURNETT – MED 92 ERITROPOESE – RESUMO •
•
Núcleo " À medida que a célula amadurece, o núcleo diminui de volume e a cromatina fica mais condensada, até o núcleo se tornar picnótico (máxima condensação), correspondendo à célula que perdeu a capacidade de se dividir. Depois, na fase de eritroblasto ortocromático, o núcleo é expulso da célula. Citoplasma " Observa-­‐se, inicialmente, o acúmulo de RNA mensageiro, extremamente basofílico. À medida que a célula amadurece, a hemoglobina vai sendo acumulada, dando ao citoplasma uma tonalidade acidófila, que nas fases intermediárias mescla-­‐se com a basofilia do RNA (estagio de eritroblasto policromatófilo) e finalmente a substitui (no estágio de eritroblasto ortocromático). RETICULÓCITO •
•
•
•
•
Aparece após a extrusão do núcleo pelo eritroblasto ortocromático; O reticulócito conserva alguns resquícios de organelas: retículo endoplasmático, ribossomos (com RNA mensageiro) e mitocôndrias. Esses resquícios são chamados de retículos; Como mantém uma certa quantidade de RNA mensageiro, o reticulócito ainda possui capacidade de síntese proteica. De fato, de 10-­‐20% da síntese de hemoglobina é feita nesse estágio. Além disso, como mantém mitocôndrias, possui certa capacidade de respiração aeróbica; " O uso de corantes supravitais, como o azul-­‐de-­‐
toluidina, revela esses restos de organelas no interior dos reticulócitos. O reticulócito recém-­‐formado permanece de 1-­‐3 dias na medula óssea, sendo em seguida liberado para a circulação. De 1-­‐2 dias depois de entrarem em circulação, eles perdem totalmente as organelas e tem seu volume reduzido. Nesse ponto, cessa a síntese proteica e a capacidade de fazer respiração aeróbica. Os reticulócitos perdem lipídios e proteínas por meio de exocitose, sendo o principal componente perdido os receptores de transferrina, que não estarão presentes na hemácia madura. " O processo final de maturação dos reticulócitos – que consiste na eliminação de grânulos sideróticos e em modificações da membrana – ocorre no baço e é chamado de culling. Em paciente esplenoctomizados, pode haver acúmulo de hemácias com anormalidade morfológica (corpos de Howell-­‐Jolly). A determinação da percentagem de reticulócitos no sangue periférico constitui importante indicador da capacidade funcional da medula óssea diante da anemia: elevação de reticulócitos indica atividade proliferativa compensatória da medula óssea, enquanto baixa nos reticulócitos indica medula hipoproliferativa. VINÍCIUS BURNETT – MED 92 REGULAÇÃO DA ERITROPOESE •
•
Dentre os vários fatores que contribuem para o controle da produção de eritrócitos, os principais são os fatores de crescimento que agem sobre as células precursoras e estimulam seu desenvolvimento e maturação, como a eritropoetina e a IL-­‐3. Eritropoetina: " É o principal fator de crescimento que atua sobre a linhagem eritroide e regula a produção de hemácias; " Trata-­‐se de uma glicoproteína de 165 aminoácidos; " Produzida principalmente no tecido renal: • 90% é produzida nas células intersticiais peritubulares renais; • 10% é produzida nos hepatócitos. " O tecido renal é altamente sensível à hipóxia e a fatores que a simulam (como a anemia). Nesses casos, o tecido renal pode induzir uma produção de eritropoetina cerca de 1000 vezes maior do que o normal. " Como atua a eritropoetina? • O hormônio liga-­‐se a receptores expressos especificamente em precursores eritroides, estimulando a sua proliferação e diferenciação, levando a um aumento da massa eritrocitária. • Regulado por mecanismo JAK – STAT-­‐5. " Como ocorre a superexpressão do gene da eritropoetina? • Três intermediários – HIF-­‐1α, HNF-­‐4 e HIF-­‐1β – interagem com um coativador transcricional (p300), formando um complexo que interage com fatores de transcrição, criando condições para ativação da transcrição gênica localizada e aumentando a produção de mRNA correspondente ao gene da eritropoetina. " Variações no nível de eritropoetina: VINÍCIUS BURNETT – MED 92 HEMOGLOBINA •
•
Nível de hemoglobina no sangue: " A concentração de hemoglobina no sangue fica baixa até os quatro anos de idade porque há um aumento na concentração de fosfato nas hemácias de indivíduos dessa idade. Isso faz com que haja um aumento na concentração de 2,3-­‐DPG, o que diminui a afinidade da hemoglobina pelo O2. Isso leva a uma maior liberação do oxigênio na periferia, o que faz com que o rim diminua a produção de eritropoetina. Tudo isso culmina na queda da produção de hemácias. “Anatomia” da hemoglobina: " É um heterotetrâmero, composto por dois pares de globinas; " Cada umas das 4 cadeias de globina possui um grupo heme, onde se liga o oxigênio; " Hemoglobina normal no adulto: •
Tipos de hemoglobina produzidos ao longo da vida: VINÍCIUS BURNETT – MED 92 "
Talassemia: • Defeito na produção de uma das cadeias de globina, que leva à anemia. • Deficiência de uma cadeia β: " Talassemia beta menor; " Geralmente assintomático. • Deficiência de uma cadeia α: " Portador assintomático; " Não é manifestada clinicamente. • Deficiência de duas cadeias β: " Talassemia beta maior (anemia de Cooley); " Indivíduo chega a nascer (ver gráfico acima. No período perinatal, a HbF é a mais importante), mas tem anemia grave quando adulto. • Deficiência de quatro cadeias α: " Hidropsia fetal; " Indivíduo não chega a nascer, pois não passa da vida fetal. HEMÁCIAS •
•
Estrutura: " Disco bicôncavo: • Proporcionalmente ao volume, possui muita membrana e pouco citoplasma ! adaptada para trocas gasosas. " Flexível: • Passagem pelos capilares. " Esferocitose: • Defeitos nas proteínas de membrana em suas interações verticais dão à hemácia um aspecto esferoide. Constitui um tipo de anemia. " Eliptocitose: • Defeitos nas interações horizontais das proteínas de membrana dão à hemácia um aspecto elipsoide. Também constitui um tipo de anemia. Metabolismo: " Via de Embden-­‐Meyerhoff: VINÍCIUS BURNETT – MED 92 •
Ciclo de vida: " Meia-­‐vida de 120 dias; " São retiradas de circulação pelo sistema monocítico-­‐macrofágico, devido a seu esgotamento metabólico e alterações degenerativas. " Como as hemácias “velhas” são reconhecidas? • Formação de agregados de proteína de banda 3 (umas das mais abundantes proteínas transmembrana das hemácias) estabilizados por hemicromos (moléculas de hemoglobina oxidadas); • Agregados seriam reconhecidos como antígenos por anticorpos IgG autólogos e complemento. " Uma vez fagocitada, a hemácia é decomposta em seus componentes: • Proteínas e fosfolipídios são digeridos; • Hemoglobina é decomposta em globina (que será metabolizada e dará origem a aminoácidos) e o heme (que libera o ferro e forma a bilirrubina). " O ferro é reaproveitado para a síntese de nova hemoglobina. Não há no organismo uma via de excreção de ferro! ANEMIA •
•
•
•
•
Anemia não é uma doença, mas sim manifestação de uma; Ocorre por um distúrbio na produção/destruição de hemácias; Como avaliar a anemia? " VCM – tamanho das hemácias; " RDW – amplitude de variação dos eritrócitos; " HCM – quantidade de hemoglobina. Tipos: " Ferropriva; " da Doença Crônica; " Megaloblástica. Causas: " Gestação; " Hipertensão portal; " Queimadas; " Vômitos; " Diarreia; " Diuréticos. VINÍCIUS BURNETT – MED 92 SEGUNDO SEMINÁRIO – POLICETEMIA VERA •
•
•
•
Policetemia: é uma condição que decorre do aumento da massa de células vermelhas do sangue; Policetemia Vera: é uma doença neoplásica clonal caracterizada pelo aumento do volume total da massa eritrocitária independentemente da ação dos mecanismos habituais de regulação da eritropoese; Ocorre por uma mutação na JAK2; Sintomas: " Prurido; " Eritromelalgia; " Trombose; " Cefaleia; " Incômodo gastrointestinal; " Vermelhidão; " Dispneia; " Hepatomegalia; " Esplenomegalia. METABOLISMO DO FERRO E DOS FOLATOS IMPORTÂNCIA DO FERRO •
•
•
•
Maturação linfocítica; Processos lisossômicos em fagócitos; " Usado como substrato para as reações. Epitélio neural; Hemoglobina. FONTES DE FERRO •
•
•
Processo de degradação da hemoglobina; A partir da mioglobina; Ferro inorgânico – por meio da alimentação (vegetais). 02/09/2011 VINÍCIUS BURNETT – MED 92 •
Observação: " O ferro ferroso férrico (Fe3+) não consegue passar do lúmen intestinal para o citoplasma do enterócito. Por isso, a Fe-­‐redutase o converte em ferro ferroso (Fe2+), e esse é mandado para dentro da célula pelo transportador DMT-­‐1. " Uma vez no citoplasma, o ferro tem dois possíveis caminhos: ou fica armazenado dentro do enterócito na forma de ferritina ou atravessa a membrana basolateral para chegar à circulação sanguínea. • Macrófagos são responsáveis por estocar ferro na forma de ferritina. " Essa passagem para o sangue é feita pela ferroportina. Porém, uma vez no sangue, o ferro ferroso não pode ser transportado, e necessita ser reconvertido em ferro férrico. Isso acontece por meio da enzima Fe-­‐oxidase (ou hefaestina). " No plasma, o ferro é transportado pela transferrina. CARREADORES DE FERRO •
•
Ferritina " Responsáveis pela estocagem do ferro; " É um heteropolímero: • 24 subunidades que se arranjam em 12 dímeros. • Monômeros podem ser do tipo H ou L: " Ferritina rica em H: obtém o ferro mais facilmente, mas tem menor avidez por ele. É encontrada principalmente no plasma. " Ferritina rica em L: obtém o ferro com mais dificuldade, mas é mais ávida por ele. É encontrada no fígado e no baço. " Armazena até 4500 átomos de ferro " Observação: • Hemossiderina é a ferritina que não mais desempenha seu papel. Transferrina " Constitui o transportador de ferro na circulação; " É produzida no fígado e nas células de Sertoli; " Carrega dois átomos de ferro; " Receptor de transferrina: • Fe ligado à transferrina (Fe2+-­‐TF) se liga ao receptor TFR1; • Complexo receptor/ligante é endocitado; • Bomba de H+ acidifica a vesícula endocitada, liberando o ferro; • Ferro é jogado para o citoplasma por ação da DMT1; • Receptor volta para a membrana e apotransferrina é liberada. VINÍCIUS BURNETT – MED 92 CONTROLE DO FERRO INTRACELULAR •
•
•
•
Nas porções não traduzidas das moléculas de RNA, existem regiões chamadas de IREs (iron responsive elements), também conhecidos como hair pins, que são reconhecidas por proteínas chamadas de IRPs (iron responsive proteins). Quando o IRE se encontra na porção 5’ da fita, como ocorre com o RNA mensageiro da ferritina, a sua ligação à IRP irá reduzir a transcrição; Quando o IRE se encontra na porção 3’, porém, a sua ligação com à IRP irá estabilizar a molécula de RNA e promover a transcrição. Isso ocorre, por exemplo, com o RNA mensageiro do receptor de transferrina. " Observação: vale lembrar que a IRP irá se ligar ao IRE quando houver uma baixa concentração de ferro no meio! IRP1 e IRP2: REGULAÇÃO DO POOL DO FERRO •
Hepcidina: " Peptídeo de 25 aminoácidos; " Produzido no fígado; " Liga-­‐se à ferroportina 1*; " Inibe a disponibilização do ferro, bloqueando a ferroportina. " Produção: • *Quando há holotransferrina (Fe2+-­‐TF) disponível, os receptores de transferrina 1 se ligam a ela, deixando o HFE para os receptores de transferrina 2. Quando isso ocorre, o receptor de transferrina 2 ligado ao HFE sinaliza uma via que terminará com a transcrição de hepcidina. • Isso não acontece quando há pouco ferro. Nesse caso, como não há Fe2+-­‐TF disponível, o receptor de transferrina 1 é quem se liga ao HFE, impossibilitando a síntese de hepcidina! VINÍCIUS BURNETT – MED 92 "
Outros fatores que alteram a produção de hepcidina: • IL6: " Aumenta a síntese de hepcidina; " Inflamação leva à diminuição da concentração de ferro. • Anemia; • Hipóxia; Diminuem a síntese de hepcidina! • Eritropoetina. DISTRIBUIÇÃO DO FERRO •
•
•
Não existe uma via de excreção de Fe! O pouco ferro que é perdido é aquele que se encontrava no interior dos enterócitos, na forma de ferritina, quando essas células foram descamadas. A única explicação para uma baixa de Fe no organismo é sangria. (é?) TERCEIRO SEMINÁRIO – ANEMIA FERROPRIVA E ANEMIA DA DOENÇA CRÔNICA QUARTO SEMINÁRIO – ANEMIA MEGALOBLÁSTICA VINÍCIUS BURNETT – MED 92 SISTEMA RETÍCULO-­‐ENDOTELIAL – MONOCITOPOESE 09/09/2011 MONOCITOPOESE •
•
•
•
•
•
•
Os monócitos provêm do mesmo precursor mieloide que os granulócitos e os eritrócitos; São as células mais antigas filogeneticamente, inicialmente apenas com o mecanismo de fagocitose; Contém vários prolongamentos que facilitam a fagocitose; Não tem grânulos definidos e seu núcleo não é lobulado; O aumento de monócitos é inespecífico; Identificam aquilo que não é próprio do organismo e fagocita; Quando migram para os tecidos, “viram” macrófagos. PROCESSO DE MATURAÇÃO •
•
•
Monoblasto " Alta relação núcleo/citoplasma: • Pouco citoplasma ! baixa síntese proteica. " Cromatina jovem e frouxa; " Núcleo indentado com nucléolo; " Citoplasma espumoso; " Basofílico. Promonócito " Presença de desmossomo: • Região clara perinuclear; • Corresponde ao local onde há maior síntese proteica; " Aparecem grânulos primários e secundários; " Discreta condensação da cromatina; " Perda do nucléolo. Monócito " Corresponde a 10% das células sanguíneas; " Grânulos não são tão bem definidos; " Núcleo não é lobulado; " Cromatina muito menos condensada (mais jovem); • Imunorregulação: ao contrário dos granulócitos, não sofrem degranulação e destruição após a fagocitose. " Citoplasma mais basofílico e espumoso; " Presença de vacúolos. VINÍCIUS BURNETT – MED 92 IDENTIFICAÇÃO IMUNOFENOTÍPICA •
Observação: " CD 15 " CD 14 Marcadores de maturidade " CD 11b " HLA-­‐DR – Apresentação de antígenos CONTROLE DA MONOCITOPOESE •
•
•
•
•
CSF-­‐1; M-­‐CSF; G-­‐CSF; TNF; IL3. •
Fatores de transcrição: " PU.1 • A falta do fator transcricional PU.1 implica na ausência de macrófagos/monócitos maduros e leva à morte perinatal. • c-­‐Jun e C-­‐EBPα • c-­‐Jun é um protoncogene que desencadeia a diferenciação monocítica; • c-­‐Jun serve de coativador do PU.1; • C-­‐EPBα se dimeriza com o c-­‐Jun e leva à down regulation do mesmo, causando o bloqueio do desenvolvimento dos macrófagos; • C-­‐EPBα também se liga ao PU.1 e leva à sua inibição de forma direta. " Lembrar que o C-­‐EPBα é um dos principais fatores responsáveis pela formação dos granulócitos. Proliferação e diferenciação dos monócitos. VINÍCIUS BURNETT – MED 92 DISTRIBUIÇÃO DOS MACRÓFAGOS •
•
Após 55 horas na medula, os monócitos perdem as moléculas de adesão que os prendiam ali e migram para o sangue, onde permanecem de 6 a 12 horas. Depois, eles vão para os tecidos (onde podem ficar até 12 meses): " Morrer; " Migração específica; " Migração inespecífica. Órgãos de destino: " Fígado: • Principal sítio de distribuição dos macrófagos; • Macrófagos são chamados de células de Kupffer. " Pulmão: • Segundo maior sítio; • Responsáveis pelo clearance de substâncias aspiradas ou produzidas nos pulmões; • Produção de elastase e colagenase, que destroem as paredes dos alvéolos. " A α1-­‐antitripsina (ausente na fibrose cística) impede a destruição do parênquima. " Baço: • Destruição das hemácias; • Formam os cordões de Billroth, juntamente com plaquetas, plasmócitos e células reticulares. " Intestino: • Função macrofágica desconhecida; • Constituem a parte periférica das placas de Peyer. " Pele: • São as chamadas células de Langerhans; • Possuem diversos prolongamentos. " Cérebro: • Chamados de micróglia. " Ossos: • Osteoclastos; • Formam sincícios (células polinucleadas); • Ligados à hematopoese (fatores de proliferação). VINÍCIUS BURNETT – MED 92 GRANULOMA •
•
•
Macrófagos delimitam a reação alérgica contra determinado microrganismo; Mantém o agente agressor preso junto a linfócitos e citosinas; " Quando há alterações no sistema imune, a bactéria é liberada ! Tuberculose. Não necessariamente destrói o microrganismo, apenas o isola. COMPONENTES DOS MACRÓFAGOS •
•
•
•
•
•
•
•
•
•
CD 4 "
"
Receptor do HIV; Macrófago é reservatório de HIV, pois o vírus infecta a célula sem destruí-­‐la. Receptores Fc " Fc = região comum dos anticorpos. Receptores do complemento; Receptores de transferrina e lactoferrina; Receptores de citosinas, interleucinas e CSF; Integrinas; " Responsáveis pela migração. Receptores de peptídeos quimiotáticos; Receptores de fatores de coagulação; Receptores de componentes de microrganismos; Receptores de corticoides. PRODUTOS DOS MACRÓFAGOS •
•
•
•
•
Lisozima, elastase, colagenase, hidrolase; ROS: substâncias reativas de O2; RNS; Lipídios bioativos; Citocinas e fatores de crescimento. FUNÇÕES DOS MACRÓFAGOS •
•
•
•
•
Clearance de restos celulares; Estoque de ferro; Fagocitose; Apresentação de antígenos: função mais aprimorada dos macrófagos; Imunomodulação: comunicação com células em volta. VINÍCIUS BURNETT – MED 92 APRESENTAÇÃO DE ANTÍGENOS – SISTEMA HLA •
•
•
•
Os antígenos leucocitários humanos constituem um “super locus” situado no cromossomo 6, que codifica diversos genes relacionados ao sistema imune humano; Os genes codificam proteínas apresentadoras de antígenos; Os antígenos apresentados são reconhecidos pelos receptores de células T (TCR); " Além do mecanismo geral de ligação entre o HLA e o TCR, cada classe de HLA apresenta regiões que servem de correceptores para essa ligação. Os HLAs são divididos em duas classes: " Classe 1: • Formado por três loci gênicos: A, B e C; • Apresentam peptídeos presentes no interior da célula em que se encontram (mas não necessariamente endógenas); • Linfócitos T citotóxicos (T-­‐CD8) reconhecem os antígenos apresentados; • Linfócitos T citotóxicos se ligam ao HLA por meio do correceptor CD8. Este se liga à cadeia α3 dos HLA de classe 1; • β2-­‐microglobulina: proteína de ancoragem que estabiliza a ligação do HLA; " β2m também se liga ao HFE, regulando a endocitose de ferro para as células do intestino. • Presente em todas as células com exceção das hemácias. " Classe 2: • Formado pelos loci DP, DQ e DR; • Apresentam antígenos de fora da célula “capturados” por meio de fagocitose; • Reconhecidos pelo linfócito T-­‐CD4; • Linfócitos T-­‐CD4 se ligam ao HLA por meio do correceptor • Presente nos macrófagos. QUINTO SEMINÁRIO – DOENÇA DO ENXERTO CONTRA O HOSPEDEIRO •
•
Condições para que ocorra: " O enxerto deve conter células imunologicamente competentes; " O receptor do enxerto deve expressar antígenos que não estão presentes no doador; " O receptor deve ser incapaz de reagir (imunologicamente) às células transplantadas. Precaução: " Escolher doador com o máximo de histocompatibilidade possível. VINÍCIUS BURNETT – MED 92 ONTOGÊNESE E DIFERENCIAÇÃO DO TECIDO LINFOIDE 23/09/2011 LINFOCITOPOESE •
•
•
Ao contrário dos mieloides, que não conseguem ter respostas diferentes a diferentes agentes agressores, os linfócitos possuem a capacidade de identificar agressores variados devido à linfocitopoese; Os linfócitos são produzidos na medula óssea e depois se diferenciam no timo (linfócitos T) e na própria medula (linfócitos B); Paul Ehrlich: “Teoria dos receptores” " Células têm receptores específicos para determinados antígenos. PROCESSO DE DIFERENCIAÇÃO DOS LINFÓCITOS B •
•
•
•
•
Linfócitos B são inicialmente produzidos no saco vitelínico; depois, durante a vida fetal, são produzidos no fígado; por fim, na medula óssea; Um dos marcadores mais precoces da linhagem B é o CD 19, que está presente até estágios intermediários do processo de maturação e é dito um marcador pan-­‐B; Na fase pro-­‐B, os precursores dos linfócitos B sintetizam a desoxinucleotidil terminal transferase (TdT), responsável por aumentar a diversidade dos linfócitos B por meio da recombinação somática; CD 10: marcador de imaturidade; CD 20: marcador de maturidade. VINÍCIUS BURNETT – MED 92 RECEPTORES DE LINFÓCITOS B (IMUNOGLOBULINAS) •
Cadeia pesada: " Gene está no cromossomo 14; " Na conformação embrionária germinativa, o gene da cadeia pesada é subdividido em quatro regiões: variável (V), de diversidade (D), juncional (J) e constante (C); • Cada uma das regiões V, D e J contém um número diferente de segmentos (ver figura acima, nos quadros mais superiores da esquerda o número aproximado de segmentos de cada região). • A região constante é específica do tipo de imunoglobulina. Assim, Cμ (C mi) dará origem a uma IgM, Cδ (C delta) dará origem a uma IgD e assim por diante. " Nas fases iniciais de diferenciação B, ocorre um rearranjo dos genes da cadeia pesada, de forma que um segmento de cada região é “escolhido”. Assim, um segmento V se combina com um segmento D, com um segmento J e com um dos segmentos constantes; • Como cada região tem diversos segmentos, o número de combinações possíveis de um segmento de cada um delas garante enorme diversidade aos receptores de linfócitos B. " A combinação obtida dá origem a um gene ativo, que codifica a síntese da cadeia pesada específica por meio da transcrição e tradução do mRNA; " Outro mecanismo que aumenta a variabilidade do processo é a ação da TdT. Essa enzima insere um número variável de novas bases de DNA na região D no momento do rearranjo gênico. • Nem todas as células serão funcionais depois desse processo, pois pode haver frameshift e não existir um códon de finalização adequado. Essas células não funcionais entram em apoptose. VINÍCIUS BURNETT – MED 92 •
•
Cadeia leve: " Existem dois genes que codificam as cadeias leves: κ e λ; • O κ se encontra no cromossomo 2 e o λ, no cromossomo 22; • O gene κ é muito mais expresso que o λ. " O processo de rearranjo é basicamente igual ao das cadeias pesadas. A única mudança é que nos genes de cadeias leves só existem regiões V e J; Depois de todas as combinações possíveis e a união de cadeias leves e pesadas, estima-­‐se que o número de linfócitos B diferentes possíveis de se formar esteja entre 1019 e 1020. MATURAÇÃO DOS LINFÓCITOS B (continuando o que estava falando antes das imunoglobulinas) •
•
•
Pro-­‐B " Primeira célula da linhagem B identificada; " Transcritos estéreis; • Correspondem ao material genético com todas as sequências correspondentes aos segmentos das regiões V, D, J e C. " Rearranjo da cadeia pesada; • Genes RAGs começam a agir no processo de recombinação das cadeias pesadas; • Exclusão alélica: nessa etapa, escolhe-­‐se um dos alelos do gene da cadeia pesada (ou do pai ou da mãe). A escolha é feita assim: o alelo que conseguir terminar uma combinação (que não leve à apoptose) primeiro será o escolhido. " Como não há cadeia leve ao final da fase de Pro-­‐B, a cadeia pesada fica acompanhada de uma chaperona, que tem a função apenas de sustenta-­‐la. Pré-­‐B " “Surge” quando o rearranjo da cadeia pesada acaba; " Reativa-­‐se o processo de rearranjo, dessa vez das cadeias leves; • A cadeia κ é preferencial; caso dê errado um rearranjo com ela, utiliza-­‐
se a cadeia λ. Em cerca de 2/3 dos casos, nenhuma das duas “funciona” e a célula entra em apoptose. " No final da fase Pré-­‐B, há dois receptores citoplasmáticos (dupla expressão de Ig): IgM e IgD ??? Linfócito B " Presença de CD 19, CD 10 e Ig; " Linfócitos T no cordão de Billroth estimulam a diferenciação do Pré-­‐B em linfócito B transicional 1. Depois, essa célula vai para o folículo primário, onde é apresentada a antígenos. As células muito reativas são eliminadas e as células que reagem de forma moderada são estimuladas a se diferenciarem em linfócitos B maduros “virgens”. VINÍCIUS BURNETT – MED 92 CENTRO GERMINATIVO •
•
Os linfócitos B maduros virgens migram para os órgãos linfoides secundários (linfonodos, pele, mucosa, placas de Peyer) e lá são apresentados a antígenos pelas células dentríticas; Os linfócitos B, então, adentram as regiões de centros germinativos; " Região escura: proliferação das células B e hipermutação somática. • Na hipermutação somática, são gerados diversos novos linfócitos B, todos diferentes entre si. Assim, alguns desses linfócitos terão maior poder de combate ao agente apresentado e outros terão uma reação mais branda contra eles. " Seleção dos linfócitos cujas mutações os tornaram mais aptos a combater o antígeno específico e indução à via de apoptose dos demais; " Linfócitos selecionados passam pelo processo de mudança de classe para se tornar mais específicos: • Splicing induzidos por citocinas e fatores de splicing para deletar/anular uma região e expressar outras Ig. " Linfócitos são liberados na circulação ou na forma de plasmócitos ou na forma de células de memória. LINFÓCITOS T •
•
•
•
•
Caracterizam-­‐se pela presença de CD 3; Nascem a partir de células comprometidas com a linhagem T no timo; " CD 44 e CD 117. Sofrem maturação no timo; Receptores de linfócitos T: " Podem ser do tipo αβ (90%) e γδ (10%); " Sofrem rearranjo semelhante às cadeias pesadas e leves dos linfócitos B; " Importantes na ligação do linfócito T com os antígenos a ele apresentados (HLA-­‐TCR); Todos os linfócitos T nascem CD8+ e CD4+. Ver abaixo como eles viram T-­‐CD8 ou T-­‐CD4. VINÍCIUS BURNETT – MED 92 •
•
•
Seleção dos linfócitos T que serão colocados em circulação: " Dentro do timo, os linfócitos T pré-­‐
formados encontram HLA tanto do tipo I quanto do tipo II (lembrar que nesse início, o linfócito é tanto CD8+ quanto CD4+); " Os linfócitos T que conseguem ligar seus TCRs ao HLA são selecionados. Os que não conseguem entram em via de apoptose. Esse mecanismo é chamado de seleção positiva. " Os linfócitos selecionados positivamente que são reativos aos antígenos apresentados sofrem apoptose. Os que não apresentam reação são liberados para a circulação. Esse processo é chamado de seleção negativa. • Por que? Lembrar que todos os antígenos apresentados nessa etapa são endógenos. Assim, deve-­‐se selecionar os linfócitos T que não tem a capacidade de destruir o próprio organismo! Linfócito T-­‐auxiliador (T-­‐CD4) " Liga-­‐se ao HLA de classe 2; " TH1 ! modula respostas por via celular; " TH2 ! modula respostas por via humoral (anticorpos); Linfocito T-­‐citotóxico (T-­‐CD8) " Liga-­‐se ao HLA de classe 1; " Estimulação pelo antígeno específico faz com que o linfócito T-­‐CD8 libere as proteínas perfurina e granzima, que vão perfurar a membrana e destruir a célula própria que está expressando um antígeno não próprio (célula própria infectada cujo HLA de classe 1 expressava um antígeno estranho encontrado em seu interior). VINÍCIUS BURNETT – MED 92 •
Células NK (natural killer) " Possui dois tipos de receptores: • Receptores ativadores do killer (KAR); • Receptores inibidores do killer (KIR). " O KAR se liga inespecificamente a várias coisas. O que impede que as células NK destruam as células próprias do organismo é a ligação dos receptores KIR com os HLA de classe 1. Quando as duas ligações são feitas simultaneamente, a célula NK não exerce sua ação. Quando apenas o KAR se liga, a célula NK entende que se ligou a uma célula estranha (pois não reconhece os HLA de classe 1) e a destrói. SEXTO SEMINÁRIO – LINFOMA DE BURKITT FIM DA PRIMEIRA PROVA VINÍCIUS BURNETT – MED 92 GRUPOS SANGUÍNEOS 07/10/2011 SISTEMA DE GRUPOS SANGUÍNEOS •
Um sistema de grupos sanguíneos corresponde a um grupo de antígenos que se relacionam e que não tem recombinações entre seus loci gênicos. IMPORTÂNCIA DO SISTEMA ABO •
•
Anticorpos são altamente efetivos (IgM); Anticorpos ocorrem de forma natural. Ou seja, não é necessária uma sensibilização prévia (como no caso do sistema Rh). ANTÍGENO H •
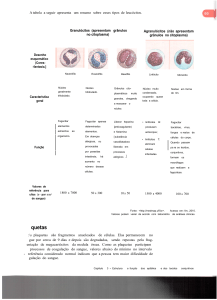
•
•
A especificidade do sistema ABO (na verdade, de todos os sistemas cujos antígenos são carboidratos) depende da molécula de açúcar incorporada a um substrato básico. Esse substrato básico é o chamado paraglobosídeo. A esse paraglobosídeo está ligada uma galactose, que pode estar ligada ou por uma ligação β(1-­‐3) ou por uma ligação β(1-­‐4). Quando a ligação é a β(1-­‐3), teremos o paraglobosídeo 1; quando a ligação é a β(1-­‐4), teremos o paraglobosídeo 2. O paraglobosídeo 1 é encontrado nas secreções, enquanto o paraglobosídeo 2 é encontrado nas hemácias. Quando se adiciona uma fucose por uma ligação α(1-­‐2) à essa galactose terminal do paraglobosídeo 2, produz-­‐se o antígeno H. essa reação é catalisada pela enzima α-­‐2-­‐
fucosiltransferase 1; " Codificada pelo gene H, presente no locus FUT1, no cromossomo 9; " Na ausência do alelo H (paciente homozigoto recessivo, hh), o indivíduo não produz a α-­‐2-­‐fucosiltransferase 1 e não expressa o antígeno H. Esses são os chamados Bombay. Quando a fucose é adicionada ao paraglobosídeo 1, o indivíduo pode expressar os antígenos A, B e “O” nas secreções. " Gene Se, no locus FUT2 produz a α-­‐2-­‐fucosiltransferase 2, que age em glicídios de secreção (saliva, trato gastrointestinal, vias aéreas). SISTEMA ABO •
Independentemente do tipo sanguíneo, todos possuem certa quantidade de antígeno H nas hemácias. A quantidade de antígeno H (“não diferenciado”) na superfície da hemácia depende da quantidade de α-­‐2-­‐
fucosiltransferase 1 e das glicosiltransferases, que convertem o antígeno H em antígeno A ou antígeno B. VINÍCIUS BURNETT – MED 92 •
•
•
•
O alelo A codifica uma glicosiltransferase que adiciona um glicídio n-­‐
acetilgalactosamina ao antígeno H; O alelo B codifica uma glicosiltransferase que adiciona um glicídio de galactose ao antígeno H; O tipo sanguíneo “O” expressa apenas o antígeno H na superfície dos eritrócitos, pois não expressa as enzimas glicosiltransferases; Manutenção do antígeno H: " Como dito, a quantidade “remanescente” de antígeno H na superfície das hemácias depende da quantidade e da “força” de conversão das glicosiltransferases. Abaixo estão os grupos sanguíneos em ordem da quantidade de antígeno H expresso (excluindo o tipo “O”): •
Bombay: " Corresponde ao tipo sanguíneo das pessoas que não possuem a α-­‐2-­‐
fucosiltransferase 1. Dessa forma, elas são incapazes de formar o antígeno H e por consequência disso, não geram nenhum outro antígeno “normal” (A1, A2, B); " Os indivíduos Bombay expressam apenas o paraglobosídeo. Assim, podem doar hemácias para qualquer grupo sanguíneo, pois ninguém possui um “anti-­‐
paraglobosídeo”; " O grande problema de um indivíduo Bombay é a confusão que se pode fazer ao tentar identificar seu tipo sanguíneo. Fazendo uma tabela convencional (abaixo), obtém-­‐se para o Bombay o mesmo resultado de uma pessoa que é do tipo O: •
O que fazer para diferenciar o Bombay do O, portanto? " Deve-­‐se fazer também o teste com um sangue sabidamente O (OHc). Nesse caso, o indivíduo O não apresentará resposta, ao contrário do indivíduo Bombay: VINÍCIUS BURNETT – MED 92 DOAÇÃO DE SANGUE •
Doação de concentrado de eritrócitos "
Observação: SISTEMA Rh •
•
•
Diferentemente do sistema ABO, cujos antígenos são carboidratos, os antígenos do sistema Rh são protéicos; Compreende 45 antígenos dos quais cinco se destacam: " D • É o antígeno mais importante do sistema Rh; • Não existe o fenótipo “d” (como se fosse um alelo recessivo). Ou o indivíduo é “D” ou não possui “D”; • Indivíduos que possuem D são ditos Rh+, enquanto que indivíduos que não possuem esse antígeno são ditos Rh-­‐. • Existem indivíduos chamados Dfracos, que apresentam uma hipoexpressão do antígeno D, embora contenham o gene devido a uma mutação no promotor do gene D. Esses indivíduos são diagnosticados como Rh-­‐, quando na verdade são Rh+ • Em alguns casos, o antígeno D é malformado, de maneira que um indivíduo D+ com essa mutação não seja mais apto a doar sangue para um indivíduo D+ “normal”, pois seus antígenos D não são iguais. São os chamados Dparciais. " C/c " E/e O locus RH, que contém os dois genes (D e CE) que codificam esses antígenos, encontra-­‐se no braço curto do cromossomo 1; VINÍCIUS BURNETT – MED 92 •
Eritroblastose fetal: " Mãe Rh-­‐ que dê à luz um bebê Rh+ pode ser sensibilizada durante o parto. Quando isso acontece, ela passa a produzir anticorpos anti-­‐D; • Vale lembrar que os anticorpos anti-­‐D não são anticorpos “naturais”; ou seja, é necessária a sensibilização para que eles sejam produzidos. Isso não ocorre, por exemplo, no sistema ABO. " Em uma gestação subsequente, na qual o feto também seja Rh+, os anticorpos anti-­‐D passarão pela placenta e causarão lise das hemácias desse feto. • Daí vem o nome eritroblastose fetal ! os eritroblastos do feto passam a ser encontrados em circulação. SISTEMA DUFFY •
•
Importante por estar relacionado com o mecanismo de entrada do Plasmodium vivax, protozoário causador da malária, nas hemácias humanas; Contém dois antígenos principais, que funcionam de maneira semelhante ao sistema ABO: a
•
" Fy ; " Fyb. O duplo negativo Fy(a-­‐ b-­‐) é extremamente raro, mas é muito comum nas regiões endêmicas de malária. Na maioria dos casos, a mutação está no promotor do gene que liga o fator de transcrição GATA-­‐1 (ver bem pra cima!), que é fundamental para a expressão de vários genes. Assim, o gene Fy não se expressa no tecido eritroide, mas a expressão da proteína Duffy é normal em outras células. SEXTO SEMINÁRIO – TRALI VINÍCIUS BURNETT – MED 92 TROMBOCITOPOESE 21/10/2011 MEGACARIÓCITO •
•
•
Célula muito grande, com citoplasma bastante granulado; Quando madura, apresenta prolongamentos, de onde surgem as plaquetas; Seu núcleo é muito grande e apresenta poliploidia como resultado da fusão de núcleos de células progenitoras devido a um processo conhecido como endomitose: " Ocorre graças a um “erro” na mitose dessas células, entre as fases de anáfase A e anáfase B; " O progenitor megacariocítico possui uma subexpressão do fator transcricional AIM-­‐1, que é responsável por gerar proteínas da família das aurora-­‐quinases; • As proteínas dessa família são essenciais na divisão celular, pois controlam a segregação dos cromossomos. " Como resultado, os megacariócitos chegam a apresentar um núcleo 128n; isso leva a uma otimização na produção de proteínas por essas células. MATURAÇÃO MEGACARIOCÍTICA •
•
•
•
•
•
A unidade formadora de colônias de granulócitos, eritrócitos, macrófagos e megacariócitos (CFU-­‐GEMM), quando apresentada a determinados fatores (ver ao lado), dá origem à BFU-­‐Meg (unidade formadora de blastos de megacariócito); A BFU-­‐Meg: " Expressa o marcador de série mieloide CD 33; " Expressa o marcador de linhagem megacariocítica CD 41; " Dá origem de 100 a 500 megacariócitos. Seguinte à BFU-­‐Meg no processo de maturação vem a CFU-­‐Meg (unidade formadora de colônia de megacariócitos). Essa expressa o CD 42a, também marcador da linhagem megacariocítica; " A CFU-­‐Meg dá origem de 4 a 16 megacariócitos. Em seguida, vem o estágio de pró-­‐megacarioblasto, que é o último estágio a se dividir; Como consequência, o estágio seguinte (megacarioblasto) é o último formado por divisão mitótica; Depois, tem-­‐se o estágio de pró-­‐megacariócito, que é seguido do megacariócito em si. Esse expressa também o CD 61 e é responsável pela formação de plaquetas. VINÍCIUS BURNETT – MED 92 REGULAÇÃO DA TROMBOCITOPOESE •
•
A trombopoetina (TPO) estimula a produção de células comprometidas com a linhagem megacariocítica, além de estimular o crescimento e a maior produção de grânulos pelos megacariócitos já existentes; Ela é produzida principalmente no fígado, mas também nos rins. A sua produção é contínua e constante; ou seja, independentemente da necessidade do organismo de produzir mais ou menos plaquetas, a produção de trombopoetina (que é quem regula isso em primeira instância) é constante. " O que explica a maior ou menor produção de megacariócitos (e de plaquetas, consequentemente)? • A trombopoetina “útil” é a que se encontra livre; • Quando encontra um receptor (na membrana do megacariócito ou da plaqueta), a TPO se liga a ele e não mais estimula a produção de células comprometidas com essa linhagem; • Assim, quando há necessidade de maior produção de plaquetas, grande parte dos receptores de trombopoetina das membranas dos megacariócitos é internalizada. • Ao lado, os gráficos mostram as conse-­‐
quências de uma trombocitopenia e de uma trombocitose no organismo: no primei-­‐
ro caso, a TPO livre aumenta para que haja formação de um maior número de plaquetas; no segun-­‐
do, ocorre o contrário: como há um excesso de megacariócitos, o TPO livre cai, de forma a regular tal processo. VINÍCIUS BURNETT – MED 92 FORMAÇÃO DAS PLAQUETAS •
•
•
•
•
A TPO estimula a formação dos megacariócitos bem como o aumento no número de grânulos no citoplasma dessas células; Diversos fatores estimulam a formação de microtúbulos no citoplasma dos megacariócitos, entre eles o NF-­‐E2; " NF-­‐E2 age sobre o TUBB1, gerando tubulina. " Pessoas que não expressam o NF-­‐E2 são ditas plaquetopênicas. • Plaquetas dessas pessoas são redondas e estão em pequeno número. Com a presença dos microtúbulos, ocorre a formação de protuberâncias do citoplasma, que resultam na árvore com as proplaquetas nos extremos dos ramos; " Os megacariócitos possuem uma reserva de membrana plasmática em seu citoplasma – o chamado sistema de demarcação de membranas (DMS). Ele é essencial para que a célula consiga emitir todos esses prolongamentos. No citoplasma dos megacariócitos existe também uma rede de proteínas de contração actina e miosina 2; " A contração dessas fibras leva ao corte e à espiralização da fibra, liberando as plaquetas do megacariócito; " Pessoas que não possuem a miosina 2 apresentam uma doença chamada anomalia de May-­‐Hegglin. • Plaquetas desses indivíduos são gigantes e estão em pequeno número (plaquetopenia). " O processo de corte das plaquetas é mediado pelo SDF-­‐1, que aumenta a resposta quimiotática dos megacariócitos de forma que esses liberem maior número de plaquetas. O corpo residual do megacariócito é então fagocitado. "
O processo de formação das protuberâncias no citoplasma do megacariócito começa em uma região chamada “polo de corrosão”. VINÍCIUS BURNETT – MED 92 ESTRUTURA PLAQUETÁRIA •
•
•
Grânulos α: " Função: • Contém proteínas ligadas diretamente ao processo de hemostasia. " Conteúdo: • P-­‐selectina; • Integrinas; • Fator de von Willebrand; • Glicoproteínas; • Fibrinogênio; • Fibronectina; • Trombospodina. Grânulos densos: " Função: • “Dizem” para outras plaquetas que elas tem que migrar para aquele lugar. " Conteúdo: • Serotonina; • ADP; • Cátions bivalentes. Sistema canalicular aberto: " Resquício do DMS dos megacariócitos; " Importante para a ativação da plaqueta. • Plaqueta ativada muda de forma: • •
Esqueleto de membrana: " Constitui a via de sinalização das glicoproteínas de membrana. VINÍCIUS BURNETT – MED 92 DINÂMICA PLAQUETÁRIA •
•
•
Vida média em circulação: " 10 dias. " Observação: • Plaquetas transfundidas ficam 7 dias em circulação. A taxa de consumo diária “basal” de plaquetas é de 7000/mℓ; A marcação com 111In permite ver que de 25% a 35% das plaquetas produzidas são retiradas de circulação imediatamente pelo baço. SÉTIMO SEMINÁRIO – PÚRPURA TROMBOCITOPÊNICA IMUNE HEMOSTASIA PRIMÁRIA •
•
28/10/2011 Hemostasia: processo de reparo de lesões celulares; Hemostasia primária: " Trata da interação que existe entre as plaquetas e o endotélio, que resulta na formação do trombo plaquetário; " A hemostasia primária é que nos faz parar de sangrar, enquanto que a hemostasia secundária (próxima aula) impede que o sangramento retorne. • Vale lembrar que as hemostasias primária e secundária ocorrem simultaneamente! ENDOTÉLIO VASCULAR E FLUXO SANGUÍNEO •
Subendotélio: " É uma matriz extracelular composta por uma série de proteínas sintetizadas pelas células endoteliais que funcionam como proteínas adesivas: • Colágeno; • Laminina; • Fibronectina; • Fator de von Willebrand; " O subendotélio é exposto para a luz do vaso quando há lesão do mesmo, o que faz com que as plaquetas possam se aderir a ele e possam formar o trombo primário. VINÍCIUS BURNETT – MED 92 •
•
Endotélio: " Em condições normais, o endotélio vascular regula o tônus vascular e garante uma superfície antitrombótica para o fluxo sanguíneo. Os principais fatores anti-­‐trombogênicos são: • Prostaglandina I2 (PGI2) e óxido nítrico (NO), que causam aumento na concentração de cAMP e cGMP no interior das plaquetas. Isso as torna menos reativas, diminuindo a agregação a outras plaquetas; " O dipiridamol é uma droga que aumenta a concentração intracelular de cAMP, inibindo a agregação plaquetária. • Expressão de CD 39 pelas células endoteliais; " O CD 39 é uma ecto-­‐ADPase, que hidrolisa o ADP liberado pelos eritrócitos destruídos e por outras plaquetas. Esse ADP é um dos responsáveis pela agregação plaquetária. • O endotélio também impede a agregação das plaquetas por ser uma barreira física entre a luz dos vasos, onde se encontram as plaquetas, e o subendotélio, onde estão os principais fatores de agregação plaquetária (colágeno e fator de von Willebrand). Fluxo sanguíneo: " O fluxo regular de sangue pelos vasos faz com que as plaquetas passem de forma ordenada, sem conseguir aderir ao endotélio. Além disso, esse fluxo “lava” os fatores protrombóticos. RECEPTORES PLAQUETÁRIOS •
•
Existem diversas nomenclaturas diferentes para os receptores plaquetários. As mais usadas são: " A baseada na eletroforese das proteínas de membrana das plaquetas. Essa classificação deu origem aos receptores com nomes de GP (glicoproteína); " A baseada na superfamília das integrinas, que são proteínas que dependem de sinalização para serem ativadas e que contém uma subunidade α e uma subunidade β. Como existem várias classificações, um mesmo receptor plaquetário pode ter mais de um “nome”. Por exemplo, o receptor GPIa/IIa também é chamado de α2β1; o receptor GPIIb/IIIa também é chamado de αIIbβ3. No quadro acima, na coluna dos receptores, todos aqueles que se encontram em uma mesma linha dizem respeito a um só receptor. VINÍCIUS BURNETT – MED 92 •
Entre os receptores plaquetários mais importantes, destacam-­‐se: " Receptor GPIIb/IIIa: • Receptor que se liga ao colágeno e é responsável por unir plaquetas próximas; " Receptor GPVI: • Responsável pela ancoragem das plaquetas; " Receptor GPIb/IX: • Liga-­‐se ao fator de von Willebrand e é o grande responsável pela “captura” das plaquetas; • A deficiência nesse receptor causa a síndrome de Bernard-­‐Soulier, caracterizada por diátese hemorrágica (sangramentos sem causa aparente). HEMOSTASIA PRIMÁRIA •
•
As etapas na hemostasia primária são: lesão, iniciação, recrutamento e perpetuação. Lesão: " A lesão expõe a matriz extracelular que forma o subendotélio, com as suas proteínas agregadoras. Destacam-­‐se o fator de von Willebrand (FvW) e fibrinogênio; " As células endoteliais, uma vez lesadas, não conseguem mais liberar PGI2 e NO e não expões mais o CD 39. Isso aumenta a agregação plaquetária; " O fluxo sanguíneo passa a ser turbilhonado, o que aumenta a probabilidade das plaquetas se ligarem ao endotélio e iniciarem o processo de formação do trombo primário. " Observação: • Também existe fator de von Willebrand em circulação. A diferença é que esse é bem menor do que o presente no subendotélio, o que não permite que as plaquetas criem o trombo apenas se ligando a ele. • O que garante que o FvW “grande” não vai estar em circulação é uma enzima chamada ADAMTS 13, que cliva o fator. VINÍCIUS BURNETT – MED 92 •
Iniciação: " O processo de iniciação se inicia com o rolamento (semelhante ao rolamento dos neutrófilos) da plaqueta pela parede do vaso; " Quando ela chega ao local lesado, o receptor GPIb/IX da plaqueta “bate” no complexo formado pelo colágeno e pelo FvW e ela é capturada; " A interação entre a plaqueta e o colágeno é feita por diversos receptores. Entre eles: • GPVI: " Faz com que a plaqueta pare de rolar; " Sinalização via fosfolipase C; " Produz TxA2 (tromboxano A2), que é um agente inflamatório, por meio da enzima COX-­‐1; • A aspirina age bloqueando a enzima COX-­‐1. " Ativa as integrinas α2β1 e αIIbβ3. • GPIb/IX: " Sinalização via tirosina kinase (Syk); " Causa expansão do esqueleto de actina da plaqueta. • α2β1 e αIIbβ3: " Causam sinalização para dentro das plaquetas. VINÍCIUS BURNETT – MED 92 •
Recrutamento: " A “monocamada” de plaquetas que se forma sobre o local lesado do endotélio no processo de iniciação não é suficiente para estancar o sangramento. Na etapa de recrutamento, mais plaquetas ativadas se acumulam acima dessa “monocamada”; " O recrutamento ocorre devido à liberação de TxA2 e de ADP pelas plaquetas que haviam se ancorado. Assim, plaquetas circulantes se ativam com mais facilidade, pois não precisam se ligar às proteínas do subendotélio; " As plaquetas podem ser ativadas por diversas vias. Entre elas, estão: • Ativação por receptores acoplados a proteína G: •
•
: Ativação pelo ADP: " Observação: o clopidogrel é uma droga que inibe o receptor P2Y12 da via de ativação plaquetária pelo ADP, impedindo a agregação das plaquetas. Ao contrário da aspirina, seu efeito não é permanente. VINÍCIUS BURNETT – MED 92 •
Perpetuação: " Para que o plug plaquetário seja estável, é necessário que haja uma forma de se ligar às plaquetas firmemente. Os responsáveis por isso são os receptores GPIIb/IIIa, que se ligam às fibras de fibrina entre as plaquetas. Como o GPIIb-­‐IIIa é um receptor da família das integrinas, é necessária uma ativação intracelular para que isso ocorra. • A droga tirofibano é um anticoagulante que inibe o GPIIb/IIIa. AGREGAÇÃO PLAQUETÁRIA •
•
•
A agregação plaquetária é medida por meio de um aparelho chamado agregômetro1; Ao lado, tem-­‐se um resultado de uma medida feita pelo agregômetro: quanto maior o grau de agregação plaquetária, mais luz é captada pelo aparelho (pois há mais “espaço livre” na amostra); Na região B da figura, mostra-­‐se o efeito de concentrações diferentes de fibrinogênio (Fg) na amostra: quanto maior a concentração, maior a agregação e, consequentemente, mais luz é captada pelo agregômetro. 1
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1516-­‐84842004000400003 VINÍCIUS BURNETT – MED 92 CAUSAS DE DISTÚRBIOS DA HEMOSTASIA PRIMÁRIA •
•
•
•
Diminuição da concentração de plaquetas (trombocitopenia ou plaquetopenia); Qualquer alteração nos receptores plaquetários; Utilização de drogas que ajam sobre as cascatas de ativação das plaquetas; Diminuição na concentração de fibrinogênio, ADP, cátions bivalentes. OITAVO SEMINÁRIO – PÚRPURA TROMBOCITOPÊNICA TROMBÓTICA •
Causa: " Anticorpos contra a ADAMTS 13. HEMOSTASIA SECUNDÁRIA 04/11/2011 CASCATA DA COAGULAÇÃO •
•
•
O modelo da “cascata” diz que a coagulação ocorre por meio da ativação proteolítica sequencial de pró-­‐enzimas (zimogênios) por proteases do plasma, resultando na formação de trombina (fator IIa) que, então, converte a molécula de fibrinogênio (fator I) em fibrina; Didaticamente, divide-­‐se a cascata da coagulação em duas vias: intrínseca (que é iniciada por componentes presentes no interior dos vasos) e extrínseca (que envolve tanto elementos do sangue quanto elementos que usualmente não se encontram expostos na luz dos vasos); " As duas vias convergem para a mesma etapa: a ativação do zimogênio X em fator Xa, que resultará na formação de trombina e na consequente conversão de fibrinogênio em fibrina. Via intrínseca: " In vivo, tem uma importância menor do que a via extrínseca, uma vez que foi constatado experimentalmente que a deficiência em um dos fatores dessa via não traz grandes prejuízos para a hemostasia do paciente; " Na via intrínseca, a ativação do fator XII ocorre quando o sangue entra em contato com uma superfície contendo cargas negativas. Tal processo é denominado “ativação por contato”. " O fator XIIa ativa o fator XI, que por sua vez ativa o fator IX. Esse se liga ao fator VIII, como será visto na via extrínseca. VINÍCIUS BURNETT – MED 92 •
Via extrínseca: " A via extrínseca da cascata de coagulação se inicia quando há exposição de uma glicoproteína chamada fator tecidual. Ela é exposta quando há lesão do vaso ou quando há alterações bioquímicas no meio (liberação de citocinas). • Fator tecidual: " Também é chamado de tromboplastina, fator III ou CD 142; " O fator tecidual (FT) é expresso em células que normalmente não estão expostas à luz do vaso, como os fibroblastos subendoteliais e nas fibras de músculo liso; • Quando ocorre uma lesão, a exposição da matriz extracelular do subendotélio expõe, além dos fatores importantes na hemostasia primária, o fator tecidual. " A liberação de TNFα e interleucina-­‐1 leva à expressão do FT pelas células endoteliais e pelos monócitos. " Em condições normais, uma pequena quantidade do fator VIIa (fator VII ativado) se encontra em circulação. Quando há exposição do fator tecidual na luz do vaso, o fator VIIa se liga a ele em uma membrana fosfolipídica e na presença de cálcio. Esse conjunto é chamado de complexo tenase extrínseco; • O complexo tenase extrínseco “recruta” os zimogênios IX e X e os ativa em IXa e Xa, respectivamente. Contudo, esse complexo possui uma eficiência muita baixa em ativar o zimogênio X; • O fator X gerado ativa o zimogênio II (protrombina), gerando o fator IIa (trombina). " Como dito, a quantidade de trombina gerada pelo complexo tenase extrínseco é mínima, mas essa pequena quantidade dará origem a mais fator Xa, como segue. " O fator IIa gerado ativa os zimogênios V e VIII, formando os fatores Va e VIIIa; • O fator VIIIa se une ao fator IXa em uma membrana lipídica, na presença de cálcio, formando o complexo tenase intrínseco; " O complexo tenase intrínseco ativa o zimogênio X, formando o fator Xa, que se liga ao fator Va gerado anteriormente. A união dos fatores Xa e Va forma o chamado complexo protrombinase; • O complexo protrombinase formará mais fator IIa, que por sua vez formará maior quantidade de fibrina. VINÍCIUS BURNETT – MED 92 "
Observação: • O complexo tenase intrínseco forma o fator Xa com uma eficiência 50 vezes superior ao complexo tenase extrínseco. VITAMINA K •
•
•
•
Alguns dos fatores de coagulação e proteínas anticoagulantes fazem parte da família serino-­‐protease. São eles: " Fator II; " Fator VII; " Fator IX; " Fator X; " Proteína S; " Proteína C. Essas proteínas contém um domínio propeptídico em suas estruturas e são liberadas na forma de prozimogênio; Para que sejam transformadas em zimogênios, essas proteínas necessitam de carboxilação pela ação da enzima carboxilase, dependente de vitamina K; " Dessa forma, a ausência de vitamina K ou a utilização de drogas antagonistas dessa vitamina (como a varfarina) impedem a formação dos fatores II, VII, IX e X, impedindo a coagulação. Como age a vitamina K? " A vitamina K altera as cargas dos zimogênios que dependem dela, deixando-­‐os negativos (parte inferior da figura ao lado); " O “problema” é que as plaquetas, quando ativadas, também apresentam suas cargas negativas para o exterior (parte superior da figura ao lado). " Como sabemos, os fatores dependentes da vitamina K formam os complexos (tenases e protrombinase) utilizando a membrana externa das plaquetas. Porém, se essas duas estruturas possuem cargas negativas, o que garante a estabilidade dessa ligação? • Forma-­‐se uma camada de íons Ca+2 entre as duas estruturas! VINÍCIUS BURNETT – MED 92 FIBRINOGÊNIO •
•
•
•
O fibrinogênio é uma glicoproteína hexamétrica, que contém pares de subunidades Aα, Bβ e γ; Quando a trombina (fator IIa) age sobre o fibrinogênio, ela retira os fibrinopeptídeos A e B das subunidades Aα e Bβ; Com isso, o fibrinogênio “vira” fibrina, e pode formar polímeros; isso é essencial para a formação do trombo plaquetário; O fator XIIIa é responsável por estabilizar esses polímeros de fibrina (na figura ao lado, fator VIIIa forma as “pontes” entre os polímeros de fibrina). SISTEMA PLASMINOGÊNIO/PLASMINA – FIBRINÓLISE •
•
•
•
A fibrinólise pode ser definida como a degradação de fibrina mediada pela enzima plasmina; Esse mecanismo é um dos responsáveis pela regulação do processo de coagulação; São conhecidos dois ativadores fisiológicos do plasminogênio: o ativador do plasminogênio do tipo tecidual (t-­‐PA) e o ativador do plasminogênio do tipo uroquinase (u-­‐PA); " Os dois ativadores promovem hidrólise de uma ponte peptídica do plasminogênio, que resulta na formação de uma serino-­‐protease ativa, a plasmina. A plasmina é a responsável por degradar a fibrina em produtos de degradação de fibrina (PDF). A fibrinólise pode ser inibida por fatores que atuem diretamente sobre a plasmina (por exemplo, o inibidor α2-­‐antiplasmina [α2-­‐AP]) ou por fatores que impeçam a ação dos ativadores do plasminogênio (por exemplo, a PAI-­‐1). ANTICOAGULANTES NATURAIS •
Além da fibrinólise, o nosso organismo regula o processo de coagulação por meio da liberação de fatores anticoagulantes. Os principais são: " Inibidor da via do fator tecidual (TFPI); " Proteína C ativada; " Antitrombina. VINÍCIUS BURNETT – MED 92 •
•
•
TFPI: "
"
O TFPI inibe a ação do complexo tenase extrínseco, que ativa os fatores IX e X; O TFPI é uma proteína produzida pelas células endoteliais que apresenta três domínios: • O primeiro se liga ao complexo VIIa/FT, inibindo-­‐o; • O segundo se liga ao fator Xa e inibe sua ação; " A ligação ao fator Xa é importante para que o TFPI exerça sua ação inibitória sobre o complexo VIIa/FT. Proteína C ativada: " A proteína C se liga ao seu receptor (EPCR) na membrana do endotélio; " Quando a trombina (fator IIa) se liga ao seu receptor também localizado no endotélio (receptor de trombomodulina – TM), ela ativa a proteína C, que “vira” proteína Ca; " A proteína Ca, então, se liga a um cofator (proteína S) e inativa os fatores Va e VIIIa, inibindo a coagulação. " Observações: • A trombina, apesar de ser considerada um grande fator coagulante, possui papel anticoagulante quando produzida em baixas quantidades! • O sistema de regulação pela proteína C ativada só acontece quando a cascata de coagulação está ativada, pois depende do fator IIa. Antitrombina: " A antitrombina é o inibidor primário da trombina, mas também exerce efeito inibidor sobre os fatores IXa, Xa, XIa e XIIa; " Além disso, a antitrombina também acelera a dissociação do complexo fator VIIa/FT e impede a sua reassociação; " As moléculas de heparan sulfato e de heparina aceleram as reações catalisadas pela antitrombina. VINÍCIUS BURNETT – MED 92 AVALIAÇÃO DA HEMOSTASIA SECUNDÁRIA •
•
•
Tempo de Protrombina (TP): " Avalia a via extrínseca da cascata de coagulação, verificando a atividade dos fatores VII, X, V, II e I; " Como é feito? • Colhe-­‐se uma amostra de plasma do paciente e retira-­‐
se os íons Ca+2 do mesmo (plasma citratado); • Adiciona-­‐se fator tecidual à amostra e a deixa incubada de 1 a 3 minutos; • Depois, adiciona-­‐se novamente o Ca+2 para que o processo de coagulação sanguínea se inicie; • Calcula-­‐se o tempo necessário para que se forme o coágulo de fibrina. O exame normal tem tempo de protrombina entre 11 e 16 segundos. " Observação: • O teste pode indicar um defeito em algum dos fatores da via extrínseca, mas é impossível saber o fator defeituoso apenas pela aplicação do TP. Tempo de Tromboplastina Parcial Ativada (TTPA): " Avalia a via intrínseca da cascata de coagulação, verificando a atividade dos fatores XII, XI, IX, VIII, X, V, II e I; " Como é feito? • Colhe-­‐se uma amostra do plasma, adiciona-­‐se kaolina, que é um ativador de superfície (ativa o fator XII) e substituem-­‐se as plaquetas pela tromboplastina parcial (que também formará o trombo, mas que não expressa o fator tecidual, de forma que a via extrínseca permaneça inativa); • Adiciona-­‐se o Ca+2 e calcula-­‐se o tempo necessário para que se forme o coágulo de fibrina. O normal é que esteja entre 30 e 40 segundos. Tempo de trombina (TT): " Indica um defeito na conversão de fibrinogênio a fibrina (deficiência do fator I); " Como é feito? •