O algoritmo genético nos ajustes de curvas de energia potencial de sistemas
diatômicos CH e NH
Gabriela R. Vaz (IC)*, Luciano Ribeiro (PQ). [email protected]
Ciências Exatas e tecnológicas, Universidade Estadual de Goiás. 459, 75001970, Anápolis, GO,
Brasil.
O objetivo principal desse trabalho é a obtenção da constantes espectroscópicas nos ajustes das
curvas de energia potencial (CEP) de um sistema diatômicos (com mais de um átomo em interação).
Para tanto foi necessário resolver a equação de Schrodinger eletrônica para varias configurações
nucleares, gerando as CEPs desse sistema.. Para resolve-la utiliza-se vários níveis de cálculo e
bases diferentes. As CEPs foram ajustadas para forma analítica do tipo Rydberg generalizada e
aproximação de Born-Oppenheier, as quais usam como base o algorítimo genético, o qual resume-se
em atrvés de um conjunto de numeros escolher e trabalhar com o que melhor se adaptar ao problema
gerado. Existe uma gama de metodolgias para otimização do processo, dentre eles o Algorítimo
genético vem se tornando uma boa opção, por dar uma melhor ideia da superfície da superfície de
resposta e por lidar com problemas muito complexos ( elevado número ode variáveis e restrições,
alem de um espaço de busca muito grande).
Palavras-chave: Constantes espectroscópicas. Método de Dunham. Algoritimo genético. HartreeFock.
Introdução
A teoria de algoritmo genético (AGs), surgiu em meados da década de 70,
constitui em uma técnica de busca e otimização, altamente paralela, inspirada no
princípio Darwiniano de seleção natural e reprodução genética. Basicamente, o que
um algoritmo genético faz é criar uma população de possíveis respostas para o
problema a ser tratado (inicializado) para depois submetê-la ao processo de
evolução, constituído em seis etapas: avaliação, seleção, cruzamento, mutação,
atualização e a finalização.
O AG é um método de busca global que não leva em consideração o
gradiente da superfície de resposta, evitando o problema de ficar confinado a
regiões de extremos locais, além do fato de fornecer informação sobre a frequência
de visitação de cada extremo. Esta última característica fornece maiores detalhes da
topografia da superfície de resposta, dando ideia das múltiplas possibilidades de
maximização e minimização.
AGs são tipicamente implementados através de simulação computacional
onde uma população de representações abstratas (cromossomos) de candidatos a
solução
(indivíduos)
de
um
problema
evolui
para
melhores
soluções
tradicionalmente, cromossomos são representados por sequencias binarias, porem
outras codificações e estruturas de dados são possíveis.
De forma geral, a evolução se inicia a partir de uma população inicial de
indivíduos e acontece em gerações. A cada geração o desempenho de cada
indivíduo é medido. Com base nisso, múltiplos indivíduos são selecionados e
modificados para formar a população da próxima geração.
É claro que o procedimento descrito acima pode ser executado de várias
formas diferentes. A descrição acima é intencionalmente abstrata. Do ponto de vista
prático, muitos modelos de AGs são desenvolvidos de forma exclusiva e especifica
para cada problema. Vale ressaltar também que desse ponto de vista a grande
maioria das pesquisas são orientadas para a utilização de AGs como ferramentas de
otimização. Em problemas de interesse dos físicos da matéria, destacam-se a
utilização de AGs nos trabalhos.
1
Material e Métodos
O conceito de curvas de energia potencial (CEP) de sistemas molecular é central
em química quântica. Do ponto de vista teórico aparece naturalmente como uma
consequência da aproximação de Born-Oppenheimer (ABO) na resolução da
equação de Schrödinger para um sistema com muitos elétrons.
2
Como pode ser observado, existe uma gama de metodologias para
otimização de processos. Dentre estes, o Algoritmo Genético vem se tornando uma
boa opção, por dar uma melhor ideia da superfície de resposta e por lidar com
problemas muito complexos (elevado número de variáveis e restrições, além de um
espaço de busca muito grande).
3
A forma analítica que utilizada nesse trabalho foi a Rydberg generalizada. Esta
forma é representada por
ai ρ
i
n
1+∑ ¿ exp (−ai ρ )
i
V R =−D e ¿
(1)
ou mais explicitamente
V ρ=−D e ( 1+a 1 ρ+ a2 ρ2 + a3 ρ3 + a4 ρ4 + … ) exp(−a i ρ).
Sendo que ρ=R−Req é o deslocamento da posição de equilíbrio
é a energia de dissociação e os
i=1
ai ¿
até
(2)
Req , De
N , onde representa os graus do
polinômio) são os coeficientes a serem ajustados pela o algoritmo genético 4. Nesse
caso usamos N = 6.
Resultados e Discussão
4.1 Sistema CH
Com a necessidade de energias eletrônicas para o sistema diátomo CH o
calculo a partir do nível de QCISD(T) com as bases 6-311++G foi feito 5. Todos os
cálculos foram realizados no software Gaussian 09 e utilizamos da infraestrutura do
grupo de Química Teórica e Estrutural de Anápolis (QTEA).
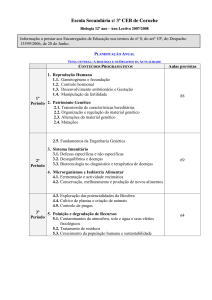
Figura 1- Energias para o Sistema CH, com níveis de calculo de QCISD(T) com as
bases 6-311++G.
Com os pontos ab initio foi construída a CEP do diátomo CH. A Figura 1
apresenta curva de energias potencial do diâtomo CH. Observe que a distancia de
equilíbrio esta em torno de 1.2 angstroms.
Os coeficientes da CEP da Figura 1 pela função analítica Rydberg, descrita
pela Equação 1, apresentados na Tabela 1 foram retirados do ajuste da Figura 2.
Tabela 1- Ajuste realizado com a Rydberg de ordem sexta para o sistema CH. A linha azul representa o ajuste, as bolas os
pontos calculados e a linha vermelha o erro realizado no ajuste.
Tabela 2- Os parâmetros ajustados com a função analítica Rydberg para os sistemas CH. As unidades em Hartree para a
dimensão de energia e Angstrom para valores de distância.
CH
a1
a2
a3
a4
a5
a6
De
R0
1.001232806987
0.755107262088
-0.070627541936
0.194956045559
-0.042119327802
0.009416931163
-2.194071052317
1.150077066831
Em temo dos valores da Tabela 1, utilizando a metodologia de Dunham
6
torna-se possível a determinação das constantes espectroscópicas. As constantes
espectroscópicas obtidas foram: frequência vibracional de equilíbrio,
constantes rotacionais
e
, para o sistema CH.
, das
A Tabela 2 estão apresentas as constantes espectroscópicas,
,
e
para o sistema CH.
Tabela 3 – Constantes espectrocópicas dos sistema CH.
CH
Be
ωe
αe
1.972
1129.5
0.0253
4.2 Sistema CN
A Figura 3 descreva a CEP para o diâtomo NH. Todo o procedimento para a
obtenção da curva de energia seguiu todos os procedimentos adotados
anteriormente.
Figura 2- Curva de energia potencial para o diatômo NH.
Para esse sistema os coeficientes ajustados com a função Rydberg estão apresentados na
Tabela X e os valores das constantes obtidas estão na Tabela y.
Tabela 4 - Os parâmetros ajustados com a função analítica Rydberg para os sistemas CN. As unidades em Hartree para a
dimensão de energia e Angstrom para valores de distância.
CN
a1
a2
a3
a4
a5
a6
De
R0
0.984573531312
0.557552856495
0.074911567278
0.095482158519
-0.013162462639
0.005284197902
-27.584138191518
1.176258197350
Tabela 5 - Constantes espectrocópicas dos sistema CN.
CN
Be
ωe
αe
1.8855
2145.5
0.00150
Considerações Finais
Nos cálculos das energias obtidas para diátomo CH e NH, Figuras 1 e 2
apresentam uma concordância no distância de equilíbrio com dados experimentais.
A próxima etapa será o ajuste da Eq. 1 aos dados obtidos das Figuras 1 e 2.
Por meio do método de Dunham calculamos as constantes espectroscópicas
harmônicas, rotacionais de equilíbrio e rotacionais de acoplamento, representadas
respectivamente por,
ω ,
Be
e
α e . Os seus valores estão em boa
concordância com valores encontrados na literatura 7.
Agradecimentos
BIC/UEG.
Referências
1.
Hardy T. ( IA : Inteligencia Artificial ). Polis, Rev la Universdad. 2001;1(2):1-23.
2.
Martins JS, Martins JBL, Silva GM. Nova Forma de Ajuste de Curvas de Energia
Potencial de Sistemas Diatômicos. Rev Process Químicos. 2012;6:39-45.
3.
Federal U, Centro S, Monografia D, Meloti I, Orientador C, Assafr C. Espalhamento de
Pósitron por Átomos e Moléculas : Aplicação ao Átomo de He e a Molécula de H 2 .
Ivan Meloti Capucho Espalhamento de Pósitron por Átomos e Moléculas : Aplicação
ao Átomo de He e a Molécula de H 2 . 2013.
4.
Antunes AWS, Ferreira Da Cunha W, Magela E Silva G, Martins JBL, Gargano R.
Dynamical properties and thermal rate coefficients for the Na + HF reaction using
genetic algorithm. Int J Quantum Chem. 2009:NA - NA. doi:10.1002/qua.22152.
5.
Esteves CS, de Oliveira HCB, Ribeiro L, Gargano R, Mundim KC. Modeling diatomic
potential energy curves through the generalized exponential function. Chem Phys Lett.
2006;427(1-3):10-13. doi:10.1016/j.cplett.2006.06.020.
6.
Dunham JL. The Energy Levels of a Rotating Vibrator. Phys Rev. 1932;41(6):721-731.
7.
Salviano LR, Esteves CS, de Oliveira HCB, Mundim KC, Ribeiro L, Gargano R. Use
of generalized exponential function to build three-dimensional reactive surfaces. Phys
A Stat Mech its Appl. 2010;389(17):3604-3612. doi:10.1016/j.physa.2010.04.031.