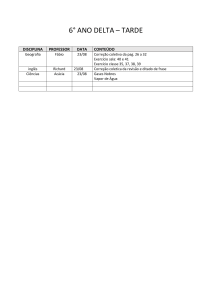
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 1
MODELAGEM TERMODINÂMICA E ESTUDO DO COMPORTAMENTO LÍQUIDOVAPOR DE SISTEMAS PRESENTES NAS ÁGUAS PRODUZIDAS EM CAMPOS DE
PETRÓLEO
Rafael H.D. Passos (UFRN)¹, Thiago C.P. Macedo (UFRN)¹, Andrielly K.S. Mota (UFRN)¹, Josinira A. Amorim
(UFRN)¹, Juliana B. Schuhli (CENPES)2, Osvaldo Chiavone Filho (UFRN)1
1
Universidade Federal do Rio Grande do Norte – UFRN, Centro de Tecnologia, Departamento de Engenharia
Química, Natal/RN, CEP 59072-970, (84) 3215-3773 r. 216, [email protected]
2
CENPES/PDP/TPAP/Grupo de Processamento, [email protected]
Na indústria do petróleo, o problema de descarte da água que provém de poços produtores é um agravante, pois
este efluente aquoso vem contaminado com sais, metais pesados e hidrocarbonetos que ao serem enviados ao
meio ambiente, sem que haja um tratamento prévio, podem provocar uma degradação da fauna e flora locais. O
objetivo deste trabalho consiste na determinação de dados de equilíbrio líquido-vapor (ELV) de uma série inicial
de misturas sintéticas e da própria água de produção. Estes dados auxiliam a análise do processo de evaporação
da água de produção para seu reuso. Os dados de ELV revelam o comportamento das misturas, que no caso da
água de produção, apresenta alta complexidade. Estes dados foram obtidos em um ebuliômetro Othmer de baixa
pressão, que recircula apenas a fase vapor. Os experimentos revelaram que os sais aumentam o ponto de ebulição
e a presença de uma segunda fase líquida orgânica abaixa o ponto de ebulição. Esse fato foi verificado com os
sistemas binários aquosos com cloreto de sódio e decano, respectivamente, bem como com o ternário. A
modelagem termodinâmica destes sistemas aquosos com sais e hidrocarbonetos requer teorias que descrevam o
efeito eletrostático, ou de longo alcance das espécies com cargas (eletrólitos), e de curto alcance, ou de
dispersão, assim como a diversidade dos compostos orgânicos, ainda que em baixas concentrações. Estes
fenômenos podem ser descritos por modelos de coeficiente de atividade como Debye-Hückel e UNIQUAC. Os
dados experimentais de ELV obtidos no laboratório e coletados na literatura são aplicados para o
desenvolvimento do modelo, assim como do seu teste para descrever os sistemas em estudo. A água de produção
apresenta curva de pressão de vapor próxima da água pura e as análises das fases em equilíbrio demonstram que
a fase vapor é pelo menos três vezes mais pobre em hidrocarbonetos do que a líquida, favorecendo o processo
evaporativo. A modelagem termodinâmica obtida dos sistemas em estudo pode ser diretamente aplicada no
processo de evaporação de tratamento das águas produzidas de petróleo.
Palavras-chave: termodinâmica, equilíbrio líquido-vapor, água de produção, comportamento das fases
1. INTRODUÇÃO
A agressão ao meio ambiente através de atividades desenvolvidas pelo homem tem sido constante. A
contínua busca pela melhor qualidade dos processos pelas empresas é resultado, principalmente, da grande carga
tributária decorrida da não obediência à legislação ambiental em vigor.
Na exploração do petróleo, a água de produção por envolver um grande volume, é o poluente mais relevante.
A água que provém de poços produtores é um efluente aquoso contaminado com sais, metais pesados e
hidrocarbonetos, o que a torna um poluente de difícil descarte, pois ao ser lançada ao meio ambiente sem que
haja um prévio tratamento, pode ocasionar danos ao ecossistema local.
O desenvolvimento de métodos adequados ao tratamento deste efluente e posterior reuso tem sido
constantemente estudados. O objetivo deste trabalho consiste na determinação de dados de equilíbrio líquidovapor (ELV) de uma série inicial de misturas sintéticas e da própria água de produção. Estes dados propiciarão
uma análise mais detalhada do processo de evaporação da água de produção.
Os dados de ELV foram obtidos em um ebuliômetro Othmer de baixa pressão, que recircula apenas a fase
vapor.
2. REVISÃO DA LITERATURA
O estudo de fases em equilíbrio é um dos principais tópicos da Termodinâmica de Soluções. Tal importância
se deve à sua incidência na indústria química, no que concerne a processos de separação, como a destilação. O
conhecimento do equilíbrio de fases é fundamental para eliminar operações onerosas no desenvolvimento
industrial, e assim economizar energia e capital.
Copyright © 2007 ABPG
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 2
Para que duas ou mais fases estejam em equilíbrio, algumas condições devem ser satisfeitas: as pressões das
fases devem ser iguais (equilíbrio mecânico), assim como as temperaturas (equilíbrio térmico) e os potenciais
químicos (equilíbrio termodinâmico dos constituintes). O potencial químico de uma espécie i em uma mistura é
definido pela relação matemática que se segue (Van Ness et al., 2000):
⎡ ∂ ( nG ) ⎤
μi ≡ ⎢
⎥
⎣ ∂ni ⎦ T , P ,n
(1)
j ≠i
Onde G é energia livre de Gibbs e n é o número de moles. O subscrito fora do colchete especifica as
propriedades mantidas constantes, nesse caso, a temperatura, a pressão, e o número de moles de todas as espécies
exceto a espécie i.
Porém, costuma-se trabalhar com a fugacidade ao invés do potencial químico para a resolução dos problemas
de ELV, por ter uma equivalência física, i.e., pseudo-pressão. Como explicitado pela equação anterior, o
potencial químico é função da energia livre de Gibbs, que por sua vez é definida em relação à energia interna e à
entropia, duas grandezas fundamentais para as quais valores mensuráveis diretos são desconhecidos. Logo, não
há valores mensuráveis diretos para o potencial químico.
A origem do conceito de fugacidade vem da seguinte equação, válida somente para espécies puras no estado
de gás ideal:
Gigi = Γi (T ) + RT ln P
(2)
Para um fluido real, pode-se escrever a equação análoga:
Gi = Γi (T ) + RT ln f i
(3)
Na Equação (3) a pressão é substituída pela fugacidade, termo corretivo para pressão devido a não idealidade
do sistema, que possui unidades de pressão. A fugacidade no estado de gás ideal da espécie pura i é igual à sua
pressão:
f i gi = P
(4)
As fugacidades podem ser determinadas através de coeficientes de fugacidade ( φ ), preferencialmente para a
fase vapor com uma equação de estado, ou de coeficientes de atividade ( γ ), usado para a fase líquida. Então, em
uma abordagem gama-phi, a equação de isofugacidade para o ELV, pode ser escrita como:
^
⎡V L ( P − Pi sat ) ⎤
yi φiV P = xi γ i Pi satφisat exp⎢ i
⎥
RT
⎣
⎦
(5)
^V
Onde yi é a fração molar do componente i na fase vapor,
φi
é o coeficiente de fugacidade na fase vapor do
componente i, xi é a fração molar do componente i na fase líquida, Pisat é a pressão de vapor do componente i
puro,
φisat
é o coeficiente de fugacidade do vapor do componente i puro, Vil é o volume do líquido saturado do
componente i puro, e R é a constante universal dos gases. O termo exponencial é chamado fator de Poynting, e
expressa os desvios da fase líquida devido ao efeito da pressão. Para pressões baixas, ou próximas à pressão de
vapor, esse termo pode ser desprezado.
O coeficiente de fugacidade do componente puro ou de misturas pode ser calculado por equações de estado.
Quanto ao coeficiente de atividade da fase líquida, a prática usualmente empregada para o seu cálculo faz uso
de modelos derivados de expressões dadas para a energia livre de Gibbs em excesso GE, que relacionam-se com
a composição e a temperatura através da expressão:
ln γ i =
1 ⎛ ∂G E
⎜
RT ⎜⎝ ∂ni
⎞
⎟⎟
⎠T , P , n j
(6)
Existem vários modelos para a energia livre de Gibbs em excesso na literatura. A certa temperatura, a energia
livre de Gibbs em excesso é função da composição do sistema, e em menor grau, da pressão, sendo que para
pressões baixas e moderadas, a dependência da pressão pode ser desprezada. Assim, os modelos adotados para a
representação do coeficiente de atividade da fase líquida ficam em função da temperatura e composição do
Copyright © 2007 ABPG
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 3
sistema. Esses modelos levam em conta a energia de interação entre as moléculas, expressa na forma de
parâmetros binários, bem como a organização das mesmas na mistura. Alguns exemplos de modelos são Wilson,
NRTL, UNIQUAC e UNIFAC.
Quanto à pressão de vapor, existem várias correlações na literatura para o cálculo da mesma. A correlação
utilizada neste trabalho foi a equação de Antoine, que é visualizada da seguinte forma:
log P sat (mmHg ) = A −
B
T (º C ) + C
(7)
As constantes de Antoine A, B e C podem ser determinadas a partir de um conjunto de dados experimentais
de pressão de vapor e temperatura tanto para um componente puro como uma mistura.
3. METODOLOGIA
Com base no que foi descrito anteriormente, sobre a modelagem termodinâmica de sistemas em equilíbrio,
pode-se aplicar para o caso particular estudado neste trabalho.
O sistema em questão avaliado é constituído por Água + Decano e Água + Decano + NaCl, pois os mesmos
possuem similaridade representativa do comportamento das propriedades físico-químicas da água de produção.
O estudo foi baseado no fato que os componentes em questão são praticamente imiscíveis entre si. Desse
modo é observada a formação de três fases, duas liquidas e uma vapor, sendo que as fases líquidas são
constituídas por componentes puros.
O descrito acima caracteriza o interessante caso de que:
x1I γ 1I
lim I
=1
x1 → 1
(8)
x2II γ 2II
=1
x2II → 1
(9)
e
lim
O resultado de tal fenômeno é descrito sucintamente nas equações de equilíbrio termodinâmico deduzidas a
seguir.
3.1 Equações Governantes
3.1.1 Sistema Água + Decano
f aV = f aI → Ya P = X aI γ aI Pasat
(10)
f DV = f DI → YD P = X DII γ DII PDsat
(11)
X aI → 1, γ aI → 1
(12)
X DII → 1, γ DII → 1
(13)
Somando as Equações (10) e (11) com as respectivas considerações, temos:
P = Pasat + PDsat
(14)
A Equação (14) representa o comportamento da solução binária em questão, onde a correlação de Antoine,
Equação (7) pode representar a dependência com a temperatura.
3.1.2 Sistema Água + Decano + NaCl
Para o sistema ternário podemos seguir com a hipótese simplificadora de imiscibilidade mútua da água e do
decano, entretanto devemos considerar o efeito do sal na fase aquosa.
Copyright © 2007 ABPG
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 4
As Equações (15) e (16) representam os equilíbrios para a água (a) e para o decano (D), sendo que para a
água temos a fase líquida aquosa e para o decano a fase liquida orgânica que de acordo com as hipóteses
simplificadoras não necessitam de correção pelo coeficiente de atividade.
f aV = f aI → Ya P = X aI γ aI Pasat
(15)
f DV = f DI → YD P = X DII γ DII PDsat
(16)
Devido ao efeito de não-idealidades proveniente da completa dissolução do sal na fase aquosa, faz-se
necessário prever tal fenômeno. O modelo utilizado neste trabalho é o UNIQUAC + Debye-Hückel, sendo que o
termo DH considera as interações eletrostáticas de longo alcance das espécies com carga.
X DII → 1, γ DII → 1
(17)
A correção pelo modelo é introduzida no cálculo da fugacidade da água, pois o sal apenas interage com a
mesma, devido a sua alta polaridade.
Somando as Equações (15) e (16) com as respectivas considerações, temos:
P = X aI γ aI Pasat + PDsat
(18)
3.1.3 Considerações sobre o modelo utilizado
FORÇA IÔNICA
Em soluções eletrolíticas a força iônica I também é comumente usada para definir uma quantidade que
expressa o conteúdo total da espécie iônica dissolvida, que em termos de simples concentrações iônicas, é
influenciada pelos valores das cargas iônicas. Esta escala de concentração tem sido aplicada em modelos de
coeficientes de atividade, inclusive em soluções diluídas onde as forças eletrostáticas são mais pronunciadas
(Pitzer, 1980), representado pela teoria de Debye-Hückel (1923).
1 Nions
I x = ∑ x j z 2j
i
2 j =1
(19)
COEFICIENTE DE ATIVIDADE
O coeficiente de atividade trabalha com as não-idealidades da solução (fase líquida e sólida). É normalmente
normalizado em referência à solução de Raoult ou de Henry.
Fazendo a solução ideal de Raoult, assume-se que os componentes interagem entre si de forma similar
quando comparadas as interações quando eles estão sozinhos, ou puros. Contudo, na verdade as misturas
apresentam forças intermoleculares facilmente diversificadas e é o coeficiente de atividade que vai levar em
consideração estas interações. Para soluções eletrolíticas as equações para o coeficiente de atividade devem levar
em consideração tanto as interações físicas, chamadas de van der Waals, como as de longo alcance, ou do tipo
eletrostática que pode ser descrita pela teoria de Debye-Hückel (1923).
Para o eletrólito dissociado o coeficiente de atividade médio iônico (γ±) pode ser representado pelas equações
somadas de UNIQUAC e Debye-Hückel (DH). Os coeficientes de atividade de cada componente, ou espécie,
são, portanto, calculados de acordo com a classe de dissociação assumida e pela combinação dos modelos
UNIQUAC e DH.
ln γ k = ln γ kUNIQUAC + ln γ kDH
(20)
A Equação (20) é utilizada para um solvente, ou um eletrólito não dissociado. Para um eletrólito a média
geométrica com os coeficientes estequiométricos da reação de dissociação é aplicada.
[
UNIQUAC + DH
γ ± = (γ cation
Copyright © 2007 ABPG
)
υ cation
(γ
)
UNIQUAC + DH υ anion
anion
]
1
(υ cation +υ anion )
(21)
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 5
O modelo UNIQUAC (Abrams e Prausnitz, 1975) consiste de duas partes, uma parte combinatorial (C) que
serve para descrever a contribuição entrópica dominante, de organização das espécies, e uma parte residual (R)
que é devido às interações entre as espécies.
ln γ kUNIQUAC = ln γ kC + ln γ kR
(22)
A parte combinatorial é determinada somente pela composição, tamanho e formato das espécies, expressa
pela seguinte equação:
ln γ kC = ln
φk
z
θ
φ
+ qk ln k + 1k − k
xk 2
xk
φk
∑x 1
j
lj =
;
j
j
Z
(rj − q j ) − (rj − 1)
2
(23)
Onde φk e θk referem-se às frações de volume e área superficial da espécie k definidas a seguir. A Equação
(23) mostra que 1j é calculado em função número de coordenação Z que é fixado igual a 10 para líquidos, e dos
parâmetros de volume rj e área superficial qj.
φk =
rk x k
∑ rj x j
θk =
;
j
q k xk
∑ qjxj
(24)
j
A parte energética, ou residual, depende das forças intermoleculares e contém os parâmetros de interação
binária ajustáveis por informação experimental. O coeficiente de atividade de uma espécie k para o termo
residual é dado pela seguinte expressão.
⎡
⎤
θ jτ kj ⎥
⎛
⎞
⎢
ln γ = qk 1 − ln⎜⎜ ∑ θ jτ jk ⎟⎟ − ∑
⎢
⎥
⎠ j ∑ θ iτ ij ⎥
⎝ j
⎢⎣
i
⎦
R
k
(25)
Onde:
⎡ u ij − u jj ⎤
⎡ aij ⎤
⎥ = exp ⎢ − ⎥
RT ⎦
⎣
⎣ T ⎦
τ ij = exp ⎢ −
u ij = u ji ; τ ij ≠ τ
ji
; a ij ≠ a
(26)
(27)
ji
A contribuição eletrostática (DH) para a energia de Gibbs em excesso é dada pela seguinte expressão, a qual
está apresentada em Pitzer (1980).
I
⎛ 4A
G E , DH
= − (∑ nk )⎜⎜ DH , x x
RT
⎝ 3b
(
⎞
⎟⎟ ln 1 + b I x
⎠
)
(28)
ADH,x é o parâmetro Debye-Hückel
A diferenciação da Equação (28) fornece a expressão do coeficiente de atividade de qualquer espécie,
solvente (zk = 0) e íons.
(
)
⎡∂ G E ,DH /(RT ) ⎤
ln γ kDH = ⎢
⎥
∂nk
⎦ T , P , n j ≠k
⎣
ln γ
Copyright © 2007 ABPG
DH
k
⎡⎛ 2 z 2
A
= − DH , x ⎢⎜⎜ k
3 ⎣⎢⎝ b
z k2 I x − 2 I x I x ⎤
⎞
⎟⎟ ln 1 + b I x +
⎥
1+ b Ix
⎥⎦
⎠
(
)
(29)
(30)
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 6
EFEITO DO SAL NOS PROCESSOS DE SEPARAÇÃO
Nesta seção descreve-se principalmente o efeito do sal na destilação.
Quando um sal está dissolvido num solvente misto consistindo de dois componentes líquidos voláteis
miscíveis, o sal pode afetar as atividades dos dois componentes voláteis através da formação de fases líquidas
associadas ou complexos. Se o sal dissolvido associa-se preferencialmente com as moléculas de um componente
do solvente comparadas com aquelas do outro, a relação da solubilidade entre os dois componentes voláteis é
alterada tal que um componente estará “salted out” mais próximo da fase vapor em relação ao outro. Em tais
casos as atividades dos dois componentes voláteis de cada uma das soluções líquidas são relativamente alteradas
de maneira que resulta numa modificação da composição da fase vapor no equilíbrio, apesar do sal não estar
presente na fase vapor.
3.2 Aparato Experimental
A montagem experimental utilizada para determinar os dados é mostrada na Figura 1. O ebuliômetro Othmer
modificado é basicamente constituído de uma célula de circulação com dispositivos de medição da temperatura e
pressão, e dispositivos auxiliares tais como manifold, traps, tanque pulmão, bomba de vácuo, agitador magnético
e regulador de voltagem. Oliveira et al. (2003) apresentam mais detalhes do dispositivo experimental.
Figura 1: Ebuliômetro Othmer
3.2.1 Reagentes
Os dados apresentados na Tabela 1 indicam os reagentes utilizados nas análises com suas respectivas purezas
e fornecedores.
Tabela 1. Substância, fornecedores, densidades e purezas para das substâncias em estudo.
Substância
Fornecedor
Água tridestilada
Decano
Cloreto de Sódio
Lab. FOTEQ
MERCK
VETEC
Densidade a 25,0°C
(g/mL)
0,9864
0,73
-
Pureza (%)
> 96
P.A.
3.2.2 Procedimento Experimental
Para assegurar que não há vazamento, submete-se o sistema a uma pressão de 300 mmHg e observa-se a
manutenção da pressão, dada pelo nível da coluna de mercúrio, por pelo menos 30 min. O dispositivo composto
de manifold, traps, tanque pulmão (20 L) e uma bomba de vácuo é usado para estabelecer a pressão constante no
sistema.
No ebuliômetro Othmer modificado coloca-se água tridestilada para a calibração do termômetro que foi
utilizado nas medidas das temperaturas de ELV dos sistemas estudados. Um agitador magnético Fisatom
(100x100 mm, Mod. 752 A, Pot. 650 Wmáx, 230) foi usado no refervedor do ebuliômetro, para misturar a fase
líquida e a fase vapor condensada retornada do amostrador, e na fase vapor condensada. O monitoramento visual
Copyright © 2007 ABPG
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 7
da ebulição, realizado para controlar a condensação parcial ou eventual superaquecimento do sistema, requer um
controle na voltagem do Variac (tipo ATV-215 M). Inicia-se normalmente com os componentes puros e
recomenda-se a determinação da faixa de voltagem ótima a ser usada para cada substância, antes de trabalhar
com as misturas. Porém, a voltagem adequada é determinada através da visualização da mistura em ebulição e
refluxo de condensado, tanto do coletor de vapor quanto da camisa externa da própria célula de equilíbrio.
O banho termostático (TE-184 TECNAL) após estar devidamente ligado e com a temperatura estabilizada em
5ºC para a água de circulação, é feito o adicionamento de gelo nos traps, que têm como finalidade condensar os
vapores arrastados pelo sistema de vácuo quando em operação, evitar perda de reagente e proteger a bomba.
Após os condensadores do ebuliômetro de Othmer estarem sob refrigeração, introduz-se a mistura em torno de
100 mL em sua câmara de mistura para se adquirir um nível adequado e reduz-se a pressão do sistema até a
pressão de trabalho. Posteriormente fecha-se a válvula de vácuo e desliga-se a bomba à vácuo (TE-581
TECNAL).
Para a calibração do termômetro, duas curvas de pressão para a água tridestilada foram obtidas, encontrandose assim uma equação para correção da pressão, que por sua vez foi adotada para corrigir a temperatura dos
demais pontos de ELV.
Solução de decano em água mantendo-se a proporção de 3,2 de água para 1,0 de decano foi preparada para
obter os dados ELV. Posteriormente, adicionou-se a esta solução cloreto de sódio até que a mesma estivesse com
a concentração de 2000ppm, representando desta forma aproximadamente as águas produzidas nos campos de
petróleo.
4. RESULTADOS
Para a validação do sistema e calibração do equipamento foi feito a curva de calibração para a água
tridestilada. A figura 1 mostra os dados de temperatura experimental confrontados com a temperatura calculada a
partir da equação de Antoine (Gmehling, 1995).
Tcorr (°C)
105
100
95
90
85
80
75
70
65
Tcorr = 0,997 Texp
R² = 0,999
65
70
75
80
85
90
95
100
105
Texp (°C)
Figura 1: Curva de calibração para a água tridestilada. Constantes de Antoine, Equação (7), do banco de dados
de Dortmund (DDB) usadas para a calibração: A = 8,0713; B = 1.730,63 e C = 233,426.
Foram obtidas curvas de pressão de vapor para o sistema decano-água com a seguinte proporção: decano (1,0
v/v) e água (3,2 v/v). Mantendo essa proporção de solvente foi adicionado 2000 ppm de sal (cloreto de sódio),
visando representar a composição da água de produção de petróleo. A figura 2 apresenta os resultados obtidos na
forma de pressão verso temperatura.
Copyright © 2007 ABPG
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 8
800
750
700
Pressão (mmHg)
650
600
550
500
450
400
350
300
250
70
75
80
85
90
95
100
105
Temperatura (oC)
Figura 2: Dados de equilíbrio líquido-líquido-vapor para o sistema água+decano+cloreto de sódio; c água
tridestilda; ¡ água + decano; água + decano +cloreto de sódio. A relação de água e decano é de 3,2:1 em
volume para todas as misturas e a concentração de sal é de 2000ppm.
Foi observado que a modelagem proposta calculando a pressão total para esse sistema de duas fases líquidas
como a soma das pressões de vapor da água e do decano representou satisfatoriamente os dados. Observou-se
também que o modelo UNIQUAC+DH representou satisfatoriamente o efeito do sal. Entretanto, o efeito do sal
no sentido de aumentar o ponto de ebulição não predominou sobre o efeito de abaixamento do mesmo dado pela
presença da segunda fase líquida. Este comportamento está de acordo com o observado anteriormente com a
própria água de produção em estudos anteriores (Mota et al., 2001).
5. CONCLUSÃO
O sistema água+decano+sal foi estudado com respeito ao comportamento de fases do tipo líquido-líquidovapor. Este sistema foi inicialmente escolhido para uma série sistemática do estudo do comportamento líquidovapor da água de produção. Os efeitos da segunda fase orgânica e da presença do cloreto de sódio na fase aquosa
foram avaliados e também descritos pela abordagem termodinâmica proposta.
6. AGRADECIMENTOS
Os autores agradecem à Universidade Federal do Rio Grande do Norte (UFRN), ao Programa de Recursos
Humanos da Agência Nacional de Petróleo (PRH-ANP-14) e ao CENPES (Centro de Pesquisas da Petrobras).
7. REFERÊNCIAS
ABRAMS, D.S. & Prausnitz, J.M., Statistical Thermodynamics of Liquid Mixtures: A New Expression for the
Excess Gibbs Energy of Partly or Completely Miscible Systems. AIChE J., 21, 116-128, 1975.
PITZER, K.S., Electrolytes From Dilute Solutions to Fused Salts. J. Am. Chem. Soc., 102, 2902-2906, 1980.
DEBYE, P. e HÜCKEL, E., “Zur Theorie der Eletrolyte I: Gefrierpunktserniedrigung und verwandte
Erscheinungem.”, Phys. Z., 24(9), 185-207, 1923.
CUNHA, G. M. A.; Evangelista Neto, A. A.; Chiavone Filho, O.; Silva, D. N.; Nascimento, C. A. O. Tratamento
preliminar da água produzida em campos de petróleo utilizando o processo foto-fenton. In: Rio Oil & Gas
Expo and Conference, 2006, Rio de Janeiro.
GMEHLING, J. Dortmund Data Bank. DDBST Software & Separation Technology, Oldenburg, Germany, 1995.
MOTA, A.L.N., Silva, D.N., Oliveira, J.D., Dantas, J.H.A., Oliveira, H.N.M., Chiavone-Filho, O. Determinação
de solubilidade mútua e equilíbrio líquido vapor para sistemas aquosos com hidrocarbonetos. In: 1º
Congresso de Pesquisa e Desenvolvimento em Petróleo e Gás, 2001, Natal.
Copyright © 2007 ABPG
4o PDPETRO, Campinas, SP
21-24 de Outubro de 2007
6.2.447-3 – 9
OLIVEIRA, H. N. M.; Lima, C. K. M., Mota, A. L. N.; Dantas Neto, A. A., Chiavone Filho, O. Projeto de
ebuliômetros de circulação da fase vapor e testes com misturas de dodecano+tween 20 e curva de destilação
de gasolina. In: 2º Congresso Brasileiro de P&D em Petróleo & Gás, 2003, Rio de Janeiro.
OLIVEIRA, H. N. M. Determinação de dados de equilíbrio líquido-vapor para sistemas hidrocarbonetos e
desenvolvimento de uma nova célula dinâmica. Setembro de 2003. p.163. Tese de doutorado. PPGEQ/DEQ,
UFRN, Natal, 2003.
REID, R.C.; PRAUSNITZ, J.M.; POLING, B.E. Properties of Gases and Liquids, 4th Edition, McGraw-Hill.
1987.
SANDLER, S.I. Chemical, Biochemical, and Engineering Thermodynamics. 4th Edition. John Wiley & Sons,
Inc. 2006. P489-572.
VAN NESS, H. C.; ABBOTT, M. M.; SMITH, J. M. Introdução à Termodinâmica da Engenharia Química. 5º
edição. Rio de Janeiro: LTC editora, 2000. p283-452.
THERMODINAMIC MODELING NAD VAPOR-LIQUID BEHAVIOR STUDY FOR
THE SYSTEMS PRESENT INTO THE PRODUCED WATER IN PETROLEUM FIELDS
In the industry of the oil, the problem of discarding of the water becomes from producing wells, is an
aggravating one, therefore this effluent contaminated with salts, heavy metals and hydrocarbons that when send
back to the environment, without a previous treatment, can provoke a degradation of the local fauna and flora.
The objective of this work consists of the determination of vapor-liquid equilibrium (VLE) data of an initial
series of synthetic mixtures and the proper water of production. These data provide a better analysis of the
evaporation process of the water of production for its reuse. The VLE data determine the mixtures behavior that
in the case of the production water, presents high complexity. These had been carried out in a Othmer
ebuliometer of low pressure, that recirculates only the vapor phase. The experiments were important to conform
that the salt increase the boiling point and the presence of a second organic liquid phase lowers the boiling point.
This was verified with the aqueous binary systems with sodium chloride and decane, respectively, as well a
ternary system. The thermodynamic modeling of these aqueous systems with salt and hydrocarbons requires
theories that describe the electrostatic or long-range effect of electrolytes, as well as the diversity of organic
components, still that in low concentrations. These phenomena can be described for models of activity
coefficient, as Debye-Hückel and UNIQUAC. The experimental data of VLE gotten in the laboratory and
collected in literature are applied for the development of the model, as well as of its test describing the systems
in study. The production water presents curve of vapor pressure next to the pure water and the analyses of the
phases in equilibrium demonstrate that the phase vapor is at least three times poor in hydrocarbons of that the
liquid, favoring the evaporative process. The gotten thermodynamic modeling of the systems in study can
directly be applied in the evaporation process treatment of produced waters of oil.
Keyword: thermodynamic, vapor-liquid equilibrium, water of production, behavior of the phases
Os autores são os únicos responsáveis pelo conteúdo deste artigo.
Copyright © 2007 ABPG