TERMODINÂMICA APLICADA
2009 Isabel Ambar
Departamento de Engenharia Geográfica, Geofísica e Energia
Faculdade de Ciências da Universidade de Lisboa
TERMODINÂMICA APLICADA
Programa
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
Conceitos básicos da Termodinâmica.
Princípio Zero da termodinâmica. Escalas de temperatura.
Gás ideal. Teoria cinética dos gases. Gases reais.
Energia, trabalho e calor. Primeiro Princípio da Termodinâmica. Aplicações.
Máquinas térmicas e frigoríficas. Segundo Princípio da Termodinâmica. Processos
reversíveis e irreversíveis. Máquina de Carnot.
Entropia. Variações de entropia em processos reversíveis e irreversíveis.
Ciclos de gás e de vapor
Energia disponível. Trabalho máximo. Irreversibilidade.
Relações termodinâmicas formais. Aplicações à termodinâmica da atmosfera e da água do
mar.
Equilíbrio de sistemas termodinâmicos.
Propriedades físicas de uma substância pura. Transições de fase. Utilização de tabelas de
propriedades.
1
TERMODINÂMICA APLICADA
2009 Isabel Ambar
1. Conceitos básicos da Termodinâmica.
1.1. A Termodinâmica e os princípios básicos
A Termodinâmica estuda a energia, as suas transformações e as suas relações com as
propriedades da matéria. Tem a ver com os sistemas macroscópicos e baseia-se num conjunto de
princípios (ou leis) que nasceram da observação experimental. Estas leis não se podem explicar a
partir de princípios mais simples e só se a experiência os vier a contradizer é que terão de ser
abandonados ou reformulados.
O princípio zero da Termodinâmica apresenta o conceito de temperatura como aferidor do
equilíbrio térmico. O primeiro princípio corresponde a uma lei de conservação da energia em
que se estabelecem as relações entre fluxos de calor, trabalho e energia. O segundo princípio
reconhece que para além da quantidade se tem de considerar também a qualidade da energia,
ocorrendo os processos espontaneamente no sentido do decréscimo da qualidade da energia. O
terceiro princípio trata das propriedades da matéria a temperaturas muito baixas e apenas
estabelece um valor de referência para a entropia, grandeza que é introduzida pelo segundo
princípio. Destas quatro leis, as primeiras a surgirem foram a 1ª e a 2ª lei (anos de 1850s) e a que
foi formulada em último lugar (1931) foi a lei zero.
1.2. História da Termodinâmica
Sadi Carnot (1796-1832), filho de um ministro de Napoleão, combateu nas vizinhanças de Paris
em 1814. Por aquilo que se seguiu, ele ficou convencido de que uma das causas da derrota de
França tinha sido a sua inferioridade industrial. E pensou que retirando a Inglaterra a máquina a
vapor retiraria o seu poder militar (as minas deixavam de poder ser exploradas e, sem carvão não
haveria ferro e portanto armamento). Carnot também compreendeu que quem aproveitasse a
potência do vapor eficientemente, seria o dono do mundo industrial e militar. O trabalho de
Carnot baseou-se na teoria do calórico que identificava calor com um fluido sem massa,
concebendo a operação de uma máquina a vapor como a de um moinho de água, mas em que o
calórico fluia da caldeira para o condensador fazendo mover as engrenagens. Assim, haveria
conservação da quantidade de calórico à medida que ele realizava trabalho. Carnot inventou o
ciclo termodinâmico que actualmente tem o seu nome (publicou em 1824 o trabalho “Réflexions
sur la puissance motrice du feu et sur les machines propres à développer cette puissance”, que
inclui a descrição do chamado ciclo de Carnot), referindo que o rendimento das máquinas
térmicas (de Carnot) é superior ao de qualquer outra máquina a funcionar entre as mesmas
temperaturas (isto acaba por corresponder a um enunciado da 2ª lei da Termodinâmica, como
veremos).
Sadi Carnot (1796-1832)
2
TERMODINÂMICA APLICADA
2009 Isabel Ambar
A ideia do calórico e da sua conservação só foi corrigida por elementos da geração nascida por
volta de 1820: Joule (1818-1889), Thomson - Lord Kelvin (1824-1907) e Clausius (1822-1888):
- James Joule (nascido em Manchester, Inglaterra, 1818) nas suas experiências dos anos
1840s, confirmou que o calor não se conserva. Mostrou que o trabalho se pode converter
quantitativamente em calor (equivalente mecânico da caloria), que trabalho e calor são
convertíveis um no outro e que o calor não é uma substância (calórico).
-
William Thomson (nascido em Belfast, Irlanda, 1824), mais tarde Lord Kelvin, entrou
para a universidade de Glasgow com 10 anos, entrou em Cambridge em 1841 onde
terminou a licenciatura em 1843 e voltou para Glasgow em 1846. Kelvin encontrou Joule
numa conferência em Oxford e ficou impressionado com a afirmação dele relativamente à
não conservação do calor. Começou então a desenvolver a ideia de que talvez o trabalho
de Carnot (conservação do calor) pudesse continuar a ser considerado mas sem
contradizer o de Joule. É o autor de um enunciado da 2ª lei da Termodinâmica e da escala
de temperatura absoluta.
William Thomson (1824-1907)
-
Rudolf Clausius (nascido em Koslin, Prussia, agora Polónia, 1822). Foi professor em
Berlim, em Zurique e em Bona, tendo desenvolvido a questão de conciliar as ideias de
Carnot e de Joule e introduzido o conceito de entropia. Num artigo de 1865, apresentou
enunciados para a 1ª e 2ª leis da Termodinâmica (a energia do universo é constante e a
entropia do universo tende para um máximo). E também especulou sobre o modo como o
calor podia ser explicado em termos do comportamento das partículas que compõem a
matéria.
3
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Rudolf
RudolfClausius
Clausius(1822-1888)
(1822-1888)
Depois da geração nascida entre 1818 e 1824, uma nova geração veio unificar a Termodinâmica e
relacioná-la com as outras correntes da ciência que estavam entretanto a desenvolver-se:
-
Josiah Williard Gibbs (1839-1903), físico norte-americano, introduziu vários princípios,
conceitos e relações (o princípio da energia mínima, o potencial de Gibbs, a relação de
Gibbs-Duhem, etc.) tendo desenvolvido os princípios da Mecânica Estatística.
Ludwig Boltzmann (1844-1906), físico austríaco, deu uma contribuição para a ligação
entre as propriedades macroscópicas da matéria e o comportamento das partículas que a
constituem, desenvolvendo os princípios da Mecânica Estatística.
Walter Nernst (1864-1941), químico alemão, chegou a uma formulação da 3ª lei da
Termodinâmica (perto do zero absoluto as reacções químicas ocorrem sem modificação
da entropia), com base em experiências de reacções químicas.
Max Planck (1858-1947), físico alemão, propôs um enunciado mais geral para a 3ª lei, em
que fixa o valor zero para a entropia no zero absoluto.
Constantin Carathéodory (1873-1950), matemático alemão, reformulou a Termodinâmica
numa base axiomática.
Carathéodory
Planck
Nernst
Boltzmann
Gibbs
1760
1780
Watt (máq. vapor)
1769
1800
1820
Carnot
1860
Joule
1880
Thomson (Kelvin)
Clausius
4
1900
1920
1940
TERMODINÂMICA APLICADA
2009 Isabel Ambar
1.3. Terminologia da Termodinâmica
Vamos agora apresentar alguns termos e conceitos que vão ser utilizados frequentemente e dos
quais convém ter uma definição científica correcta porque muitos deles aparecem na linguagem
corrente com sentidos por vezes diferentes.
-
-
-
Sistema termodinâmico – região macroscópica limitada por uma fronteira (real ou
abstracta).
Vizinhança do sistema – região fora do sistema e que pode interagir com ele.
Universo termodinâmico – conjunto sistema+vizinhança.
Parede do sistema – fronteira, real ou imaginária, do sistema; em geral, os sistemas estão
submetidos a restrições impostas pelas suas paredes, e cada uma dessas condições
denomina-se ligação (pode haver ligações internas e externas).
Parede adiabática – parede que impede qualquer fluxo de energia térmica (calor) entre o
sistema e a vizinhança.
Parede diatérmica – parede que permite o fluxo de energia térmica (calor) entre o sistema
e a vizinhança.
Parede permeável (impermeável) – parede que permite (impede) o fluxo de matéria entre
o sistema e a vizinhança (Nota: uma parede adiabática é sempre impermeável mas o
inverso não é verdadeiro).
Parede semi-permeável – parede que permite apenas o fluxo de certas substâncias
químicas entre o sistema e a vizinhança.
Parede rígida (móvel) – parede que impõe um volume constante para o sistema (permite a
variação do volume do sistema).
Sistema fechado (aberto) – sistema envolvido por uma parede impermeável (permeável).
Sistema isolado - sistema envolvido por uma parede rígida e adiabática.
Variáveis do sistema – propriedades que caracterizam o sistema do ponto de vista
macroscópico. Exemplos: pressão (p), volume (V), temperatura (T), quantidade de
matéria, energia interna, etc. Mas nem todas as variáveis são independentes (p. ex., para
os gases a baixas pressões, pV = nRT). As variáveis independentes, às quais podemos
atribuir valores arbitrários, designam-se por parâmetros de estado; as que não são
independentes, i.e., as que são função dos parâmetros de estado, são as funções de estado.
A variação de uma função de estado num processo depende apenas dos estados inicial e
final mas não do processo entre estes dois estados. As relações entre as diferentes
variáveis de um sistema designam-se por equações de estado.
Variáveis extensivas – variáveis que dependem da massa do sistema. São aditivas no
sentido em que o seu valor no sistema é a soma dos seus valores em qualquer conjunto de
subsistemas nos quais o sistema se decomponha. Exemplos de variáveis extensivas são: o
volume (V), a quantidade de matéria (expressa, por exemplo, pelo número de moles).
Variáveis intensivas – variáveis que não dependem da massa do sistema. Exemplos são a
temperatura (T) e a pressão (p).
Variáveis conjugadas – duas variáveis, uma extensiva (X) e a outra intensiva (Y), são
conjugadas entre si se o produto YdX corresponder a uma quantidade infinitesimal de
energia. Por exemplo, pdV tem dimensões de energia (neste caso, energia mecânica).
Sistema simples – é um sistema fechado, homogéneo e isotrópico macroscopicamente,
electricamente neutro, quimicamente inerte, sem ligações internas e que não é actuado por
campos eléctricos, magnéticos, gravitacionais ou por binários de forças externas.
5
TERMODINÂMICA APLICADA
2009 Isabel Ambar
-
-
Processo termodinâmico – transformação de um estado de equilíbrio para outro, variando
as propriedades do sistema. A série de estados pelos quais o sistema passa durante o
processo denomina-se percurso.
Processo cíclico – processo cujos estados final e inicial coincidem.
Processo infinitesimal – processo em que as variáveis do sistema sofrem uma modificação
infinitesimal, em que o sistema não sai praticamente do equilíbrio.
Processo quase-estático – processo finito formado por uma sequência de estados
intermédios de equilíbrio ligados por sucessivos processos infinitesimais.
Processo reversível – é uma sucessão de processos infinitesimais que se pode inverter em
cada passo mediante uma modificação infinitesimal da vizinhança. No final seria possível
inverter o processo voltando o sistema e a sua vizinhança ao mesmo estado inicial
respectivo. Todos os processos reversíveis são quase-estáticos (i.e., todos os estados
intermédios são de equilíbrio) mas o contrário não é verdade (i.e., pode haver processos
quase-estáticos irreversíveis)
Processo irreversível quase-estático – é uma sucessão de estados de equilíbrio em que não
é possível inverter o processo voltando o sistema e a vizinhança ao estado inicial.
Reservatório de calor – é um sistema com uma massa tão grande que é capaz de absorver
ou ceder uma quantidade ilimitada de calor sem sofrer variações das suas propriedades
termodinâmicas (nomeadamente, da temperatura).
2. Princípio Zero da Termodinâmica. Escalas de temperatura
Princípio Zero da Termodinâmica
A sensação de quente ou de frio e a noção de temperatura fazem parte do nosso dia-a-dia através
dos nossos sentidos. Mas os nossos sentidos podem ser enganadores (p. ex., uma caixa de cartão
e uma “couvette” de gelo, retirados do congelador ao mesmo tempo, provocam sensações de frio
muito diferentes, apesar de estarem à mesma temperatura. Porquê?).
Vamos considerar que dois objectos estão em contacto térmico um com o outro se houver trocas
de calor entre eles. O equilíbrio térmico ocorre quando deixa de haver trocas de calor entre dois
objectos em contacto térmico um com o outro.
Na Termodinâmica, o conceito de temperatura aparece associado ao equilíbrio térmico e ao
Princípio Zero que se enuncia do seguinte modo:
Se dois sistemas A e B estão separadamente em equilíbrio térmico com um terceiro sistema C,
então A e B estão em equilíbrio térmico entre si.
Existe uma grandeza escalar, denominada temperatura, que é uma propriedade intensiva dos
sistemas termodinâmicos em equilíbrio, tal que a igualdade de temperatura é a condição
necessária e suficiente de equilíbrio térmico. Dois objectos em equilíbrio térmico um com o outro
estão à mesma temperatura. E dois objectos a temperaturas diferentes não estão em equilíbrio
térmico entre si.
6
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Os termómetros são instrumentos que medem a temperatura de um sistema. Todos os
termómetros utilizam a variação de uma propriedade física com a temperatura: (i) volume de um
líquido; (ii) comprimento de um sólido; (iii) pressão de um gás a volume constante; (iv) volume
de um gás a pressão constante; (v) resistência eléctrica de um condutor (a baixas temperaturas
utiliza-se fio de platina e a temperaturas altas um semicondutor); (vi) côr de uma peça de metal;
(vii) a força electromotriz de um par de metais (com uma junção metálica a uma temperatura de
referência e uma segunda junção à temperatura que se pretende medir: termopar).
Escalas de temperatura
Um termómetro pode ser calibrado pondo-o em contacto térmico com sistemas que se mantêm a
temperatura constante. Por exemplo, a mistura de água e gelo em equilíbrio térmico à pressão
atmosférica normal (definida como tendo uma temperatura de zero graus Celsius - 0ºC) ou a
mistura de água e vapor em equilíbrio térmico à pressão atmosférica normal (definida como
tendo uma temperatura de 100ºC). No entanto, termómetros baseados na expansão de um líquido
calibrados deste modo mas utilizando líquidos diferentes (p.ex., álcool e mercúrio) podem
apresentar discrepâncias entre eles porque as propriedades de expansão térmica das duas
substâncias são diferentes. Além disso, um termómetro de mercúrio não pode medir temperaturas
abaixo do ponto de congelação deste (-39ºC) e um termómetro de álcool não mede temperaturas
acima do ponto de vaporização deste (85ºC). Em termodinâmica, é muito conveniente ter uma
escala de temperaturas que seja independente das propriedades de qualquer substância, escala que
é denominada por escala termodinâmica de temperatura.
Notas
No séc. XVII foi desenvolvido um termómetro de vidro com um líquido mas não havia nenhum padrão para as
temperaturas. Cada cientista tinha a sua própria escala, muitas vezes baseadas em diferentes pontos de referência. No
princípio do séc. XVIII, foram apresentadas escalas universais de temperatura baseadas em pontos fixos (tais como a
temperatura da mistura de gelo e água, o ponto de ebulição da água, etc.) por diversos físicos: Daniel Gabriel
Fahrenheit (1686-1736), Anders Celsius (1701-1744), e René-Antoine Ferchault de Réaumur (1683-1757).
Fahrenheit (físico, alemão) – inventou o 1º termómetro de álcool (1709) e o 1º termómetro de mercúrio (1714);
introduziu a escala de temperaturas Fahrenheit (1724). Celsius (físico e astrónomo, sueco) – introduziu uma escala
de temperaturas centígrada (1742); a designação de escala Celsius (ºC) foi adoptada em 1948 numa conferência
internacional de pesos e medidas. William Thomson, Lord Kelvin (matemático e físico, inglês) – introduziu a escala
Kelvin (1848)
Consultar os seguintes sites da net: http://es.rice.edu/ES/humsoc/Galileo/Things/thermometer.html)
http://inventors.about.com/library/inventors/blthermometer.htm
Num termómetro de gás a volume constante, a propriedade física cuja variação com a
temperatura serve para medir esta, é a pressão. O gás (geralmente hidrogénio ou hélio) está
contido numa ampola em contacto com um manómetro de mercúrio. O volume de gás é mantido
constante elevando ou baixando o reservatório de mercúrio. A altura da coluna de mercúrio que
obriga o gás a manter o volume permite conhecer a pressão que é exercida pelo gás. Mas esta
pressão é proporcional à temperatura (ver Figura seguinte), desde que o volume se mantenha
constante. A experiência mostra que as leituras do termómetro são praticamente independentes do
tipo de gás utilizado, desde que a pressão deste seja baixa e a temperatura bem acima do ponto a
7
TERMODINÂMICA APLICADA
2009 Isabel Ambar
partir do qual o gás se torna líquido. Se se extrapolar as linhas de variação de p com T, obtidas
experimentalmente, para a região de temperaturas negativas, a pressão tende para zero quando a
temperatura atinge o valor -273,15 ºC. Esta é a base da escala de temperaturas Kelvin à qual
corresponde o valor 0 K.
p
dados medidos
•
extrapolação
- 273,15
•
•
•
•
•
•
0
•
•
Gás A
Gás B
Gás C
T (ºC)
T = a + b p, sendo a e b constantes empíricas
Nos primeiros termómetros de gás utilizou-se o ponto de fusão do gelo e o ponto de ebulição da
água para a respectiva calibração. Mas como estes pontos são difíceis de reproduzir
experimentalmente, adoptou-se uma nova escala de temperaturas baseada num único ponto: o
ponto triplo da água (temperatura à qual coexistem em equilíbrio água líquida, vapor de água e
gelo) o qual corresponde a 0,01ºC à pressão (p) de 4,58 mm de mercúrio. Nesta nova escala, o
ponto triplo corresponde a 273,16 K. Esta é a escala termodinâmica de temperaturas no sistema
SI e a unidade SI de temperatura termodinâmica é o Kelvin (definido como 1/273,16 da
temperatura do ponto triplo da água). A temperatura de 0 K é referida como o zero absoluto (em
1989, conseguiram-se atingir temperaturas de 0,000000002 K). Em Física Clássica, a energia
cinética das moléculas de um gás seria nula no zero absoluto porque aí a pressão do gás (que é
proporcional à energia cinética das moléculas) é nula. Mas isso não é verdade e a Mecânica
Quântica mostra que as moléculas nesse estado têm uma quantidade finita de energia cinética.
A temperatura na escala Kelvin é definida por um termómetro de gás ideal a volume constante
como:
⎛
⎞
⎜ p ⎟
⎟K
T = 273,16 ⎜
⎜⎜ p ponto ⎟⎟
⎝ triplo ⎠
A escala Celsius foi inicialmente definida fazendo corresponder o valor zero à temperatura do
gelo fundente à pressão atmosférica e o valor 100 à temperatura de ebulição da água à mesma
pressão (esta escala começou por se chamar escala Centígrada).
Há outras duas escalas de temperaturas que são normalmente utilizadas nos EUA e na GrãBretanha, a escala Fahrenheit e a escala Rankine. Na escala Fahrenheit, a temperatura de fusão
da água é 32ºF e a temperatura de ebulição é 212ºF. A temperatura em graus Rankine (R) é
definida arbitrariamente como 1,8 vezes a temperatura em Kelvin (K).
8
TERMODINÂMICA APLICADA
2009 Isabel Ambar
As relações entre as diferentes escalas são as seguintes:
Celsius
T (ºC) = T (K) - 273,15
T (ºF) = (9/5) T (ºC) + 32ºF
Δ T (ºC) = ΔT (K) = (5/9) ΔT (ºF)
T (R) = 1,8 T (K)
Kelvin
Fahrenheit
Rankine
671,67
Tebul água
100,00
373,15
212,00
0,01
273,16
32,02
491,69
0,00
273,15
32,00
491,67
0,00
- 459,67
0,00
- 273,15
Ponto triplo água
Tfusão água
Zero absoluto
3. Gás ideal. Teoria cinética dos gases. Gases reais.
Gás ideal
Vejamos as propriedades de um gás de massa m contido num recipiente de volume V à pressão p
e temperatura T. Estas grandezas estão relacionadas entre si e a equação correspondente
denomina-se equação de estado e é, em geral, muito complicada. No entanto, se o gás estiver a
uma pressão baixa (baixa densidade) esta equação de estado simplifica-se. O comportamento de
um gás com densidade baixa corresponde muito aproximadamente ao de um gás ideal.
Considere-se um gás ideal contido num cilindro com um êmbolo mantendo-se constante a massa
do gás. Experiências realizadas mostraram que quando o gás é mantido a temperatura constante a
sua pressão é inversamente proporcional ao volume (lei de Boyle-Mariotte: pV = constante se a
temperatura for constante) e que, a baixas pressões, o volume é directamente proporcional à
temperatura quando a pressão é mantida constante (lei de Gay-Lussac: V = Vo [1+α (T-To)]).
Estas observações podem ser traduzidas na seguinte equação de estado para um gás ideal:
p V = n Ru T
onde n é o número de moles do gás, Ru é uma constante e T a temperatura absoluta (em K). À
medida que a pressão se aproxima de zero, a grandeza pV/nT tende para um valor constante e
igual para todos os gases e por isso Ru é denominada a constante universal dos gases. Em
unidades SI (p em Pascal, V em m3), Ru tem o valor:
Ru = 8,31 J/mol.K
Se a pressão vier expressa em atmosferas (1 atm = 1,01325x105 Pa) e o volume em litros (1 L =
10-3 m3), então Ru = 0,0821 L.atm/mol.K (usando este valor, verifica-se que o volume ocupado
por 1 mole de qualquer gás à pressão atmosférica e a 0ºC é 22,4 L).
9
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Nota
Uma mole de moléculas de uma substância é a massa dessa substância que contém 6,022x1023
moléculas. Uma mole de átomos de uma substância é a massa dessa substância que contém
6,022x1023 átomos. Este número 6,022x1023 é a constante de Avogadro (NA). O número de moles
(n) de uma substância relaciona-se com a sua massa (m) através da expressão n = m/M, onde M
é a massa molar (ou a massa de uma mole) da substância. A massa molar de um elemento,
expressa em gramas, é numericamente igual ao número que exprime a massa atómica desse
elemento (p. exº., a massa molar do oxigénio atómico, O, é 16 g mol-1; a massa molar do
oxigénio molecular, O2, é 32 g mol-1).
Como o número total de moléculas (N) é igual ao número de moles (n) multiplicado pela
constante de Avogadro (NA), também podemos exprimir a lei dos gases em função do número
total de moléculas (N):
pV = nRuT = N/NA RuT = N kB T, onde kB (= Ru/NA) é a constante de Boltzmann (kB = 1,38x1023
J K-1).
m
R u T , podemos exprimir a lei dos gases ideais em termos do volume
M
1
específico (v, volume por unidade de massa): p v = R u T = R T
M
pv = RT
R
onde v é o volume específico e R = u é uma constante para cada gás (diferente de gás para
M
Notando que pV =
gás).
Nota
Conversão de unidades de pressão
1 bar = 105 Pa
1 Torr = 133,322 Pa (1 Torr equivale a 1 mm de mercúrio)
1 atm = 101325 Pa
1 psi = 6894,76 Pa (esta unidade – “pounds per square inch” – ainda é, por vezes, utilizada)
Teoria cinética dos gases
Quando falámos das propriedades de um gás ideal referimo-nos a variáveis macroscópicas tais
como a pressão, o volume ou a temperatura. O que vamos ver agora é que essas propriedades
podem ser descritas numa escala microscópica se considerarmos a matéria como um agregado de
moléculas. Vamos aplicar de um modo estatístico as leis de Newton do movimento a um
conjunto de partículas e considerar apenas o comportamento molecular de gases, nos quais a
interacção entre moléculas é mais fraca do que nos líquidos ou nos sólidos.
Na teoria cinética dos gases, consideram-se as moléculas como esferas rígidas a moverem-se de
um modo aleatório, colidindo elasticamente entre elas e com as paredes do reservatório. As
moléculas não têm outro tipo de interacção entre elas para além das colisões e não se deformam.
10
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Este modelo só é adequado para gases monoatómicos em que o único modo de energia é a
energia cinética de translação; para moléculas mais complexas em que há também rotação e
vibração das moléculas, temos de modificar esta teoria.
3.2.1. Modelo molecular de um gás ideal
Vamos considerar que:
- O número de moléculas é grande e a separação média entre elas é grande comparada com as
suas dimensões. Neste caso, podemos desprezar o volume das moléculas comparado com o
do reservatório.
- As moléculas obedecem às leis do movimento de Newton mas, no seu conjunto, movem-se
aleatoriamente (i.e., cada molécula pode mover-se em qualquer direcção e a qualquer
velocidade) e a distribuição das velocidades não varia no tempo apesar das colisões entre
moléculas.
- As moléculas colidem elasticamente (i.e., a energia cinética total e o momento linear total
conservam-se) umas com as outras e com as paredes do reservatório.
- As forças entre moléculas são desprezáveis excepto durante a colisão.
- O gás considerado é uma substância pura (i.e., todas as moléculas são idênticas).
Vamos deduzir a expressão da pressão de N moléculas de um gás ideal contido num volume V,
que será um cubo de lado d.
→
Considere-se uma molécula (de massa m) com velocidade v (componentes vx, vy, vz) que colide
com uma parede tal como está indicado na figura. Depois da colisão elástica, a componente x do
momento linear passa a ser (–mvx), e portanto a variação do momento linear da molécula é:
∆px = - m vx – (m vx) = - 2 m vx
Como há conservação do momento linear total (molécula+parede), então a variação do momento
linear da parede será (2 m vx).
-vx
vx
A força média exercida pela molécula sobre a parede no tempo ∆t (tempo entre 2 colisões com a
mesma parede) está directamente relacionada com a variação do momento linear da parede:
F1 ∆t = ∆p = 2 m vx
11
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Para que a molécula colida duas vezes com a mesma parede, terá de percorrer uma distância 2d
2d
ao longo da direcção x e o tempo que decorre entre essas duas colisões é ∆t =
. Então a força
vx
F1 será:
2mv x mv 2x
F1 =
=
Δt
d
A força total exercida sobre a parede por todas as moléculas será a soma das forças
correspondentes a N moléculas:
N
m N
m
F = ∑ Fi = ∑ v 2xi =
N v 2x
d i =1
d
i =1
em que v 2x é o valor médio do quadrado da velocidade de N moléculas na direcção x.
Considerando agora uma molécula cujas componentes da velocidade são vx, vy e vz, o quadrado
da sua velocidade é dado pela relação: v 2 = v 2x + v 2y + v 2z e portanto o valor médio de v2 para
todas as moléculas no recipiente está relacionado com os valores médios dos quadrados das
componentes:
v 2 = v 2x + v 2y + v 2z
Mas como o movimento é completamente aleatório, v 2x = v 2y = v 2z e portanto v 2 = 3v 2x .
Então, a força total sobre a parede é dada pela expressão:
N ⎛ m v 2 ⎞⎟
F= ⎜
3 ⎜⎝ d ⎟⎠
A pressão total sobre a parede é obtida dividindo a força total pela área da parede (d2):
p=
F 1 ⎛⎜ N m v 2
=
d 2 3 ⎜⎝ d 3
⎞ 1⎛ N⎞
⎟ = ⎜ ⎟ m v2
⎟ 3⎝ V ⎠
⎠
2 ⎛ N ⎞⎛ 1
2⎞
⎜ ⎟⎜ m v ⎟
3 ⎝ V ⎠⎝ 2
⎠
A pressão é proporcional ao número de moléculas por unidade de volume e à energia cinética
média da molécula. Temos então, através deste resultado, uma ligação entre uma grandeza
macroscópica – a pressão – e uma grandeza associada à estrutura microscópica. Aquela expressão
mostra que a pressão aumenta com o número de moléculas e com a energia cinética média das
moléculas, a qual, como veremos mais adiante, aumenta com a temperatura.
p=
Notas:
- Se queremos aumentar a pressão de um pneu, metemos ar no pneu.
- Não devemos mandar aferir a pressão dos pneus quando acabámos de andar muito tempo com o
carro. Porquê?
12
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Actividade
Consultar sites da net:
http://comp.uark.edu/~jgeabana/mol_dyn/KinThI.html
http://ucdsb.on.ca/tiss/stretton/chem1/gases9.html
Interpretação molecular da temperatura
Vamos agora examinar o significado de temperatura do ponto de vista molecular.
Vimos que a equação de estado de um gás ideal (que é baseada em dados experimentais) se pode
exprimir como pV = N kB T onde kB é a constante de Boltzmann. Mas a partir da expressão da
2 ⎛1
⎞
pressão, deduzida com o modelo molecular de um gás ideal, temos pV = N⎜ m v 2 ⎟ . Então,
3 ⎝2
⎠
igualando as duas expressões, vem:
2 ⎛1
2⎞
T=
⎜ mv ⎟
3k B ⎝ 2
⎠
A temperatura é uma medida directa da energia cinética molecular média. Rearranjando esta
equação:
1
3
3
v 2 = k BT
ou
mv 2 = k BT
m
2
2
1
1
1
Como v 2x = v 2 , então m v 2x = k B T e o mesmo tipo de expressão se obteria para as outras
3
2
2
duas componentes da velocidade. Quer dizer que cada grau de liberdade (i.e., para cada modo
independente pelo qual a molécula pode ter energia) translacional contribui com uma mesma
1
quantidade de energia ( k B T ) para o gás. Isto é um caso particular do teorema da equipartição
2
da energia da Mecânica Estatística.
A energia translacional total de N moléculas de gás é dada por N vezes a energia média por
molécula:
1
3
3
E = N( m v 2 ) = Nk B T = nR u T
2
2
2
visto que kB=Ru/NA e n=N/NA.
A velocidade média quadrática das moléculas é dada pela expressão:
3R u T
3R u T
3k B T
=
=
v mq = v 2 =
m
mN A
M
onde M é a massa molar (que é igual ao produto da massa de cada molécula – m – pelo número
de moléculas existentes numa mole – NA). Vemos que, a uma dada temperatura, as moléculas
mais leves movem-se mais rapidamente, em média, do que as mais pesadas (ver tabela junta).
Gás
H2
He
H2O
CO2
Massa molar (g mol-1)
2,02
4,0
18
44
13
vmq (ms-1) a 20ºC
1902
1352
637
408
TERMODINÂMICA APLICADA
2009 Isabel Ambar
3.2.2. Calores específicos
Calor específico de um gás ideal monoatómico
O calor necessário para elevar a temperatura de n moles de um gás de Ti para Tf depende do
processo entre os estados inicial e final. Considerando então diferentes processos que levam o gás
de um mesmo estado inicial para um mesmo estado final, a variação da energia interna (∆U) será
a mesma para todos eles. Mas pela primeira lei da Termodinâmica, ∆U = Q + W, e como o W
difere de processo para processo (num diagrama pV, a área sob a curva que representa o
processo) então Q também será diferente para os diferentes processos.
Consideremos então dois processos que ocorrem mais frequentemente: processo a pressão
constante (isobárico) e processo a volume constante (isocórico). Vamos definir os calores
específicos molares associados a estes processos do seguinte modo:
Q = n Cp ∆T (a pressão constante)
Q = n Cv ∆T (a volume constante)
Vimos anteriormente que a temperatura de um gás é uma medida da energia cinética média de
translação (do centro de massa) das moléculas do gás, não se incluindo a energia de vibração ou
de rotação. Considerando um gás ideal monoatómico (1 átomo por molécula; p.exº., He, Ne, Ar),
toda a energia cinética é de translação. Toda a energia recebida por um gás monoatómico num
recipiente vai aumentar a energia cinética de translação dos átomos. A energia térmica total de N
moléculas de um gás ideal monoatómico é:
3
3
N kB T = n Ru T
2
2
Note-se que a energia é função exclusiva da temperatura.
U≡E=
(a) Considere-se a transferência de calor para um gás ideal monoatómico num processo a volume
3
constante (W = 0), Q = ∆U = n R u ΔT . Todo o calor transferido vai aumentar a energia interna
2
(e a temperatura) do gás. Como num processo a volume constante Q = n Cv ∆T, então igualando
as duas expressões de Q, vem:
3
R u = 12,5 J mol -1K −1 para um gás ideal monoatómico
2
A variação de energia interna de um gás ideal pode ser expressa na forma:
∆U = n Cv ∆T
Se as variações forem infinitesimais, no limite vem:
Cv =
dU = n Cv dT
Cv =
1 dU
n dT
(b) Vamos agora considerar um processo a pressão constante em que se dá a mesma variação de
temperatura ∆T que foi considerada no caso do processo a pressão constante. O calor transferido
14
TERMODINÂMICA APLICADA
2009 Isabel Ambar
é dado pela expressão Q = n Cp ∆T, mas agora vai haver também trabalho realizado pelo gás
(porque o volume aumenta): W = - p ∆V.
Pela 1ª lei virá:
∆U = Q + W = n Cp ∆T – p ∆V
Como a energia interna de um gás ideal é função exclusiva da temperatura, e a variação ∆T é a
mesma que considerámos para o processo a volume constante, podemos escrever ∆U = n Cv ∆T.
Por outro lado, p ∆V = n Ru ∆T (pela equação de estado) e portanto:
n Cv ∆T = n Cp ∆T - n Ru ∆T
Cp – Cv = Ru
Esta relação é válida para qualquer gás ideal. O calor específico molar a pressão constante de um
gás ideal é maior que o calor específico molar a volume constante. Isto é lógico porque no caso
de um processo isocórico todo o calor é utilizado no aumento da energia interna, visto que não há
trabalho realizado pelo gás, enquanto que num processo isobárico, alguma da energia térmica é
transformada em trabalho realizado pelo sistema. No caso de sólidos ou de líquidos não há
grande diferença entre estes dois calores específicos porque num processo a pressão constante a
expansão é pequena.
3
No caso de um gás monoatómico, C v = R u , e portanto:
2
5
C p = R u = 20,8 J mol -1K -1
2
A razão entre estes calores específicos (a pressão e a volume constante) é uma grandeza
adimensional denominada coeficiente adiabático, γ:
Cp
γ =
Cv
No caso de um gás ideal monoatómico γ = 5/3 = 1,67.
Calor específico de um gás ideal mais complexo. Equipartição da energia
No caso de gases mais complexos (em que se tem de considerar as contribuições dos modos
3
vibracionais e rotacionais), já não se verifica C v = R u e por isso γ ≠ 1,67.
2
De facto, a energia térmica de um gás inclui contribuições dos movimentos de translação, de
vibração e de rotação das moléculas. Dado que estes dois últimos movimentos podem ser
activados por colisões, eles estão ligados ao movimento de translação. A Mecânica Estatística
mostra que para um número grande de partículas obedecendo às leis de Newton, a energia
disponível é, em média, comparticipada igualmente por cada um dos graus de liberdade (número
de meios independentes pelos quais uma molécula possui energia).
Consideremos então um gás diatómico em que as moléculas têm a forma de um haltere. Neste
modelo, o centro de massa da molécula pode mover-se nas direcções x, y e z. Além disso, a
molécula pode rodar em torno de 3 eixos perpendiculares entre si mas só se consideram 2 graus
de liberdade rotacionais relativos aos dois eixos que são perpendiculares ao eixo que une os
átomos. Então, há 5 graus de liberdade (3 associados com o movimento de translação e 2 com o
1
de rotação). Como cada grau de liberdade contribui, em média, com k B T de energia por
2
molécula, então a energia total de N moléculas é:
15
TERMODINÂMICA APLICADA
2009 Isabel Ambar
1
1
5
5
U = 3N( k B T ) + 2N( k B T ) = Nk B T = nR u T
2
2
2
2
O calor específico molar a volume constante pode então ser obtido do seguinte modo:
1 dU 5
Cv =
= Ru
n dT 2
E como Cp – Cv = Ru e γ=Cp/Cv, vem:
7
7
C p = R u ~29,1 J mol-1 K-1
γ = = 1,40
2
5
Estes resultados concordam bastante bem com grande parte dos resultados experimentais (ver
tabela seguinte), apesar de não terem sido contabilizados os graus de liberdade correspondentes
aos modos vibracionais (que seriam dois, associados respectivamente às energias potencial e
cinética das vibrações). Os dados experimentais sugerem que algumas das moléculas diatómicas
(p. ex., H2 e N2) não vibram à temperatura ambiente mas outros (p.ex., Cl2) vibram. Para
moléculas com mais de 2 átomos o número de graus de liberdade aumenta resultando em calores
específicos maiores, o que está de acordo com os dados experimentais.
Cp (J mol-1 K-1)
He
Ar
Kr
20,8
20,8
20,8
H2
N2
O2
Cl2
28,8
29,1
29,4
34,7
CO2
SO2
H 2O
37,0
40,4
35,4
Cv (J mol-1 K-1)
Gases monoatómicos
12,5
12,5
12,3
Gases diatómicos
20,4
20,8
21,1
25,7
Gases poliatómicos
28,5
31,4
27,0
Cp-Cv
γ=Cp/Cv
8,33
8,33
8,49
1,67
1,67
1,69
8,33
8,33
8,33
8,96
1,41
1,40
1,40
1,35
8,50
9,00
8,37
1,30
1,29
1,30
Exercício sugerido
Demonstre que num processo adiabático reversível de um gás ideal as seguintes relações são
válidas:
pVγ = constante
TVγ−1 = constante
Sugestão: considere dU = n Cv dT (variação da energia interna de um gás ideal só depende da
variação da temperatura) e dU = đQ + đW = đW = - pdV. Então, igualando estas 2 expressões de
dU: n Cv dT = - p dV. Diferenciando a equação de estado do gás ideal: p dV + V dp = n Ru dT
R
Eliminando dT entre estas duas equações: p dV + V dp = - u p dV
Cv
Substituindo Ru = Cp-Cv dividindo tudo por pV:
⎛ C p − C v ⎞ dV
dV dp
dV
dp
dV
⎟
= (1 − γ )
ou
+γ
=0
+
= −⎜⎜
⎟
V
p
V
p
V
⎝ Cv ⎠ V
Integrando, vem ln p +γ ln V = constante ou pVγ = constante
16
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Vimos que o teorema da equipartição da energia consegue explicar alguns aspectos do calor
específico mas não explica a variação deste com a temperatura. Por exemplo, o Cv do hidrogénio
5
7
é R u entre 250 K e 750 K, mas aumenta para cerca de R u para temperaturas acima de 750
2
2
K. Isto sugere que há vibrações a altas temperaturas. Por outro lado, para temperaturas muito
3
menores que 250 K, o valor é R u , sugerindo que a molécula só tem energia translacional a
2
baixas temperaturas.
As dificuldades em explicar estes fenómenos através do teorema da equipartição da energia
devem-se ao facto de a mecânica clássica ser inadequada quando se aplica a sistemas
moleculares. Só um modelo baseado na mecânica quântica seria inteiramente satisfatório.
Calor específico de sólidos
Os calores específicos dos sólidos em geral decrescem não linearmente com o decréscimo da
temperatura aproximando-se de zero à medida que a temperatura absoluta tende para zero. A altas
temperaturas (>300 K), os calores específicos molares dos sólidos tendem para um valor de 3Ru
(~25 J mol-1 K-1) como diz a lei de DuLong-Petit (para os sólidos, o produto do calor específico
pela massa molar é constante).
O calor específico de um sólido a altas temperaturas pode ser explicado pelo teorema da
equipartição. Para pequenos deslocamentos de um átomo em torno da sua posição de equilíbrio,
cada átomo realiza um movimento harmónico simples nas direcções x, y e z. A energia associada
com o movimento vibracional consiste em energia cinética (associada à velocidade dos átomos) e
energia potencial (associada à posição dos átomos). Na direcção x será então:
1
1
E x = mv 2x + kx 2
2
2
As energias na direcção do eixo dos yy e na do eixo dos zz são semelhantes. Então cada átomo do
sólido tem 6 graus de liberdade e portanto tem uma energia vibracional média de
1
6 × k B T = 3k B T . A energia total de N átomos é:
2
U = 3 N kB T = 3 nRuT
O calor específico molar de um sólido a volume constante é:
1 dU
Cv =
= 3R u
n dT
Este resultado está de acordo com a lei de DuLong-Petit.
A falta de concordância entre este modelo e os dados experimentais a baixas temperaturas devese à incapacidade da mecânica clássica para explicar alguns aspectos dos movimentos
moleculares.
3.2.3. Distribuição de velocidades moleculares
A distribuição da velocidade das moléculas de gás em equilíbrio térmico está representada na
figura junta, sendo Nv a função de distribuição de Maxwell-Boltzmann. Se N for o número total
de moléculas, então o número de moléculas com velocidades entre v e v+dv é dado por Nvdv e a
17
TERMODINÂMICA APLICADA
2009 Isabel Ambar
N v dv
. Esta fracção corresponde à
N
probabilidade de que uma molécula tenha uma velocidade entre v e v+dv.
A expressão da função de distribuição de velocidades de N moléculas de gás é:
2
m
N v = 4π N(
) 3/2 v 2 e − mv /2k BT
2π k B T
onde m é a massa de uma molécula de gás.
fracção de moléculas com velocidades entre v e v+dv é
vmp
v
vmq
(adaptado de Serway, 4ª edição)
A velocidade média v é um pouco menor do que a velocidade média quadrática vmq. A
velocidade mais provável vmp é a velocidade para a qual a curva de distribuição tem um máximo.
A partir da expressão de Nv pode-se determinar essas três grandezas:
v mq = v 2 = 3k B T/m = 1,73 k B T/m
v = 8k B T/π m = 1,60 k B T/m
v mp = 2k B T/m = 1,41 k B T/m
Comparando curvas de distribuição de velocidades para diferentes temperaturas, elas desviam-se
para à direita (i.e., para valores de v maiores) com o aumento da temperatura, tornando-se mais
largas e abrangendo um maior intervalo de velocidades. A forma assimétrica da curva deve-se ao
facto de que a menor velocidade possível é zero enquanto que o limite superior clássico é infinito.
A distribuição de velocidades moleculares de um gás, para além da temperatura, também depende
da massa da molécula. Para uma dada temperatura, a fracção de moléculas com velocidades
excedendo um dado valor aumenta à medida que a massa aumenta.
18
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Curvas para N=105
moléculas de azoto
Nv
(moléculas/
m s-1)
(adaptado de Serway, 4ª edição)
A distribuição de velocidades de moléculas de um líquido é semelhante à de um gás. O fenómeno
da evaporação de um líquido pode então ser compreendido: as moléculas do líquido com maiores
velocidades conseguem atravessar a superfície do líquido e abandoná-lo mesmo a temperaturas
muito abaixo do ponto de ebulição. Essas moléculas que escapam do líquido por evaporação têm
energia suficiente para vencer as forças atractivas das moléculas da fase líquida. Então as
moléculas que ficam nesta fase têm uma energia cinética média mais baixa e fazem com que a
temperatura do líquido baixe. É por isso que a evaporação é um processo de arrefecimento.
3.2.4. Percurso livre médio
As moléculas colidem entre si porque não são pontos geométricos e portanto não se deslocam em
linha recta. A distância média entre colisões denomina-se percurso livre médio. O percurso das
moléculas individuais é aleatório.
Vamos obter uma estimativa do percurso livre médio de uma molécula de gás. Supondo que as
moléculas são esferas de diâmetro d, duas moléculas só colidem se os seus centros estiverem a
uma distância menor que d. Isto pode ser considerado equivalente a ter uma molécula com um
diâmetro 2d e as outras serem pontos geométricos.
d
2d
Num intervalo de tempo t, uma molécula com velocidade igual à velocidade média v , percorre a
distância v t. Nesse mesmo intervalo de tempo, uma molécula com um diâmetro equivalente 2d
19
TERMODINÂMICA APLICADA
2009 Isabel Ambar
varre um cilindro com uma secção recta πd2 e comprimento v t (portanto com volume πd2 v t). Se
nV for o número de moléculas por unidade de volume, então o número de moléculas no cilindro é
(πd2 v t nV). Como a molécula de diâmetro 2d colide com todas as moléculas neste cilindro no
tempo t, o número de colisões é πd2 v t nV.
O percurso livre médio l é a distância média entre colisões e portanto é igual à distância média
percorrida a dividir pelo número de colisões no tempo t:
l =
vt
2
πd vt nv
=
1
π d 2n v
O número de colisões por unidade de tempo – frequência de colisão – é dado por f = π d 2 v n v .
O inverso é o tempo médio entre colisões – tempo livre médio.
Nesta análise considerámos estacionárias as moléculas no cilindro. Se o movimento dessas
moléculas for tido em conta, os resultados correctos são:
l =
1
f=
2
2 πd nv
2 π d2 v n v = v / l
Gases reais
3.3.1. Modelo molecular de um gás de van der Waals
Até agora considerou-se que os gases obedeciam à equação de estado dos gases ideais, (pV =
nRuT) o que é uma boa aproximação para os gases reais a temperaturas e pressões normais. Na
dedução da equação de estado dos gases ideais desprezou-se o volume ocupado pelas próprias
moléculas e as forças intermoleculares. Vamos ver agora o efeito de não desprezar estes dois
aspectos.
No caso de um gás ideal, se a temperatura se mantiver constante, a variação da pressão com a
variação do volume corresponde a uma curva hiperbólica (pV=cte) num diagrama pV. Mas se se
tratar de um gás real, as curvas experimentais diferem de hipérboles a não ser para temperaturas
muito elevadas.
As razões para este comportamento dos gases reais têm justamente a ver com o volume das
moléculas e as forças entre elas. À medida que a pressão aumenta num sistema gasoso, o volume
ocupada pelas moléculas pode-se tornar uma parte significativa do volume total e as forças de
atracção entre moléculas tornam-se importantes. Van der Waals (1873) propôs as seguintes
modificações à equação de estado dos gases ideais.
Se o volume do recipiente for V e o volume ocupado pelas moléculas for nb, então o volume
disponível para o gás é V-nb, em que b é uma constante para cada gás e n é o número de moles.
Para uma dada quantidade de gás, quanto menor for o volume do recipiente maior é a fracção
deste ocupada pelas moléculas. Então, em vez da equação dos gases ideais, teríamos:
nRu T
R T
p=
= u
V − nb V
−b
n
20
TERMODINÂMICA APLICADA
2009 Isabel Ambar
No que diz respeito às forças intermoleculares, estas são tanto mais importantes quanto mais
próximas estiverem as moléculas. A atracção entre moléculas altera as trajectórias destas
(encurva-as) aumentando o tempo de translação. Isto diminui a frequência de colisões nas
paredes, resultando num decréscimo da pressão exercida sobre estas. Este decréscimo é
proporcional à força atractiva que se exerce sobre cada molécula que se aproxima da parede e
também ao número de moléculas que se aproximam da parede por unidade de área. Qualquer
⎛n⎞
destas duas grandezas é, aproximadamente, proporcional à densidade de partículas no gás ⎜ ⎟ e
⎝V ⎠
⎛n⎞
portanto teremos um factor correctivo proporcional a ⎜ ⎟
⎝V ⎠
2
A pressão resultante é reduzida de
⎛ 2 ⎞
⎜V ⎟
⎝
⎠
um factor ⎜ n a ⎟ , em que a é uma constante. Daqui resulta a equação de estado de van der
2
Waals:
p=
Ru T
n2
− 2a
V
−b V
n
ou na forma
2 ⎞
⎛
⎜ p + n a ⎟ ⎛⎜ V − b ⎞⎟ = Ru T
⎜
⎠
V 2 ⎟⎠ ⎝ n
⎝
Em termos de volume molar (v) a equação de van der Waals tem a forma seguinte:
⎛
a ⎞
⎟⎟ (v − b ) = Ru T
⎜⎜ p +
v2 ⎠
⎝
Para cada gás, as constantes a e b são empíricas e são escolhidas de modo a ajustar a equação aos
dados experimentais. Na tabela seguinte indicam-se valores destas constantes para várias
substâncias no estado gasoso.
Substância
a
(J. m3mole-2)
b
(m3mole-1)
Ar
0,1358
3,64x10-5
Dióxido de carbono (CO2)
0,3643
4,27x10-5
Azoto (N2)
0,1361
3,85x10-5
Hidrogénio (H2)
0,0247
2,65x10-5
Água (H2O)
0,5507
3,04x10-5
Amónia (NH3)
0,4233
3,73x10-5
Hélio (He)
0,00341
2,34x10-5
Freon (CCl2F2)
1,078
9,98x10-5
21
TERMODINÂMICA APLICADA
2009 Isabel Ambar
À medida que a pressão diminui, os termos correctivos tornam-se desprezáveis. Empiricamente
observa-se que, à medida que a pressão aumenta, o termo de correcção que mais rapidamente se
torna importante é o da pressão. A equação de van der Waals tem um problema: as constantes a e
b são, na realidade, dependentes da temperatura e, portanto, os seus valores têm de ser
determinados empiricamente para diferentes domínios de p e T.
3.3.2. Outras equações de estado de gases reais
Devido à complexidade das forças intermoleculares, o comportamento de um gás real não pode
ser rigorosamente descrito por uma simples equação de estado como a de Van der Waals, mas os
aspectos básicos que dela se depreendem estão correctos. Com temperaturas muito baixas, as
moléculas com baixa energia atraem-se entre si e o gás tende a liquefazer-se. Se se aumentar a
pressão acelera-se a liquefacção. Com temperaturas elevadas, a energia cinética média é
suficientemente grande para vencer as forças de atracção entre moléculas e portanto as moléculas
não se agregam e a fase gasosa mantém-se.
A equação de estado de Beattie-Bridgeman, proposta em 1928, baseia-se em 5 constantes
determinadas empiricamente:
p=
a
v
RuT
v
2
(1 −
c
vT
3
)(v + B) −
A
v2
b
v
em que A = A o (1 − ) e B = B o (1 − ) , sendo v o volume específico molar.
A equação de estado de Benedict-Webb-Rubin, proposta em 1940, aumentou para oito o número
de constantes empíricas:
γ
R T
C
bR u T − a aα
γ − v2
1
c
p = u + (B o R u T − A o − o )
(1 +
+
+
+
)e
v
T2 v2
v3
v 6 v 3T 2
v2
A figura seguinte (curvas isobáricas num diagrama T-v) mostra a percentagem de erro
correspondente às três equações de estado referidas, no caso do azoto (% erro:
v tabela − v equação
v tabela
Ref.: Thermodynamics. An Engineering Approach. Y. A. Çengel & M. A. Boles).
22
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Vemos que o erro aumenta com o aumento da pressão e que, de um modo geral, o erro é maior no
caso da equação de van der Waals do que no caso das outras duas equações de estado.
A equação de estado de uma substância também pode ser expressa como uma soma de termos do
tipo:
p=
R u T a(T) b(T) c(T) d(T)
+
+
+
+
+ ...
v
v2
v3
v4
v5
Equações deste tipo chamam-se equações de estado do virial e os coeficientes a, b, c, d, … são os
coeficientes do virial. Estes podem ser determinados experimentalmente ou teoricamente a partir
da mecânica estatística.
4. Energia, calor e trabalho. Primeiro Princípio da Termodinâmica. Aplicações.
Sabemos que a energia não pode ser criada ou destruída mas apenas transformada de uma forma
para outra. O primeiro princípio da Termodinâmica pode ser considerado como uma
generalização do princípio da conservação da energia mecânica. Há três grandezas em jogo: a
energia interna, o trabalho e o calor. Qualquer delas tem dimensões de energia mas tanto o
trabalho como o calor só podem ser considerados como meios de transferência de energia.
4.1. Formas de energia
A energia pode existir em diversas formas: térmica, mecânica (cinética, potencial), eléctrica,
magnética, química e nuclear. A energia total (E) de um sistema é a soma destas diferentes
formas de energia. Em Termodinâmica, o foco principal é a variação da energia total e não o
valor desta e portanto pode-se fazer corresponder a um valor nulo a energia total de um sistema
num determinado ponto de referência que seja conveniente. A variação da energia total de um
sistema é independente desse ponto de referência.
Geralmente em Termodinâmica, consideram-se as várias formas de energia de um sistema
divididas em dois grupos: macroscópico e microscópico. As formas macroscópicas de energia
correspondem às que o sistema, como um todo, possui relativamente a algum referencial exterior:
energia cinética (associada ao movimento do sistema relativamente a um referencial) e energia
potencial (resultado da elevação do sistema num campo gravitacional). As formas microscópicas
de energia estão relacionadas com a estrutura molecular do sistema e com o grau de actividade
molecular, sendo independentes de qualquer sistema de referência exterior. A soma de todas as
formas microscópicas de energia designa-se por energia interna do sistema. Este termo e o seu
símbolo (U) apareceu pela primeira vez com os trabalhos de Clausius e Rankine. A energia
interna corresponde à soma das energias cinéticas e potenciais das moléculas. No caso de um gás,
as moléculas movem-se no espaço com uma certa velocidade a que corresponde uma energia
cinética translacional. Os átomos de uma molécula poliatómica rodam em torno de um eixo e a
energia associada a esta rotação é a energia cinética rotacional. Mas também podem vibrar em
torno do centro de massa comum e a energia associada é a energia cinética vibracional. No caso
dos gases, a energia cinética é principalmente translacional e rotacional mas, para altas
temperaturas, o movimento vibracional torna-se importante. Os electrões e outras partículas no
23
TERMODINÂMICA APLICADA
2009 Isabel Ambar
núcleo do átomo também têm energia cinética rotacional (rotação em torno do núcleo) e de spin
(rotação em torno do seu eixo).
A energia interna está também associada com as várias forças de ligação entre as moléculas, entre
os átomos dentro de uma molécula e entre as partículas dentro do átomo e do seu núcleo. As
forças que unem as moléculas umas às outras são mais fortes nos sólidos e mais fracas nos gases.
Se for transferida para as moléculas de um sólido ou de um líquido uma quantidade de energia
suficiente, as moléculas conseguem vencer estas forças moleculares e passar ao estado gasoso,
dando-se uma transição de fase. A energia interna associada à fase de um sistema é designada por
energia latente. A energia interna associada às ligações atómicas numa molécula corresponde à
energia química. Durante uma reacção química, algumas ligações químicas são destruídas e
outras são formadas e, como resultado, há variação da energia interna. As forças nucleares,
dentro do núcleo do átomo, são muito maiores do que as forças que ligam os electrões ao núcleo
e a energia associada a elas designa-se por energia nuclear.
A energia interna é uma função de estado do sistema (i.e., uma função dos parâmetros de estado).
A energia interna é a energia de um sistema que esteja estacionário (sem movimento de
translação e de rotação), e inclui a energia nuclear, a energia química, a energia de compressão
(como no caso de uma mola comprimida ou esticada) e a energia térmica. A energia térmica é a
fracção da energia interna que varia quando a temperatura do sistema varia. A transferência de
energia térmica é causada pelas diferenças de temperatura entre o sistema e a sua vizinhança.
As únicas formas de transferência de energia associadas a um sistema fechado são o trabalho e o
calor. No caso de um sistema aberto (ou volume de controlo), também pode haver trocas de
energia associadas ao fluxo de massa.
4.2 Transferência de energia sob a forma de calor
Até aos princípios do século XIX, considerava-se que havia transferência de uma substância
denominada calórico nos processos que se davam ao pôr dois sistemas, inicialmente a
temperaturas diferentes, em contacto, acabando ambos por ficar a uma temperatura intermédia
entre as respectivas temperaturas iniciais. O sistema a temperatura mais alta teria “mais calórico”
que o outro a temperatura mais baixa. Hoje em dia, dizemos que houve transferência de energia
sob a forma de calor. A direcção da transferência de energia é sempre do corpo a temperatura
mais elevada para o corpo a temperatura mais baixa. Calor é então uma transferência de energia
entre dois sistemas (ou entre um sistema e a sua vizinhança) devida a uma diferença de
temperaturas. Calor é energia em trânsito, não é uma propriedade do sistema!
Se o sistema estiver envolvido por uma parede adiabática, ele não poderá trocar calor com a
vizinhança. É importante notar que pode haver uma variação de temperatura do sistema sem ter
havido transferência de energia térmica (ou calor): pode ter havido transferência de energia sob a
forma de trabalho. Por exemplo, um gás num cilindro com paredes adiabáticas, ao ser
comprimido por um êmbolo, vai aquecer e a sua energia térmica aumenta (mesmo sem ter havido
transferência de calor através das paredes e do êmbolo).
24
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Nota
Antes de se ter compreendido que o calor é uma forma de transferência de energia, definia-se
calor em termos das variações de temperatura produzidas no sistema e definia-se caloria (cal)
como a quantidade de calor necessária para aumentar a temperatura de 1 g de água de 14,5º para
15,5ºC. Hoje em dia a unidade utilizada para a transferência de calor é o Joule (1 cal ≡ 4,186 J).
Mas quando nos referimos ao valor energético dos alimentos geralmente ainda usamos a Caloria
(com C maiúsculo) que corresponde a 1 kcal.
Nota
Conversão de unidades de energia
1 erg = 10-7 J
1 BTU = 1,055x103 J (esta unidade – British Thermal Unit – ainda é muito utilizada na indústria)
Há várias formas de transferir calor para um sistema: por condução, por convecção e por
radiação. A condução pode ser considerada como uma troca de energia cinética entre moléculas
em colisão. A taxa à qual o fluxo de calor por condução se dá, depende do gradiente de
temperatura.
Na convecção, a substância aquecida move-se (p. exº, o ar aquecido por um radiador numa sala).
A convecção pode ser natural, se o movimento for gerado por diferenças de densidade (ar quente
sobe) ou forçada se o movimento da substância aquecida for forçado (por exemplo, por uma
ventoinha ou por uma bomba).
A radiação provém da energia electromagnética que todos os corpos a uma temperatura diferente
do zero absoluto emitem. Um corpo negro (ou absorvente ideal) absorve toda a energia que nele
incide e emite o máximo possível para qualquer corpo com a mesma temperatura, dimensões e
forma.
Calor específico
Quando se transfere calor para um sistema (sem haver trocas de energia sob a forma de trabalho),
a sua temperatura geralmente sobe (dizemos “geralmente” porque há excepções, como durante as
transições de fase em que o calor fornecido ao sistema é utilizado para modificar a estrutura
molecular e não para elevar a sua temperatura). A quantidade de calor necessária para elevar de
1K a temperatura de uma dada massa de substância denomina-se capacidade térmica (C). Se a
quantidade δQ de energia térmica for transferida para um sistema, a temperatura deste varia de
acordo com a seguinte relação: δQ = C δT
O calor específico (c) refere-se à massa unitária da substância:
δQ
c=
m δT
Então, a energia térmica Q, transferida entre um sistema de massa m e a sua vizinhança,
correspondendo a uma variação de temperatura de Ti para Tf, será dada por:
Tf
Q = m ∫ c dT
Ti
25
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Em princípio, o calor específico depende da temperatura mas se puder ser considerado constante
no intervalo de temperaturas daquele integral, então podemos escrever simplesmente:
Q = m c (T f − Ti )
O calor específico, no caso de um sistema simples, tem valores diferentes conforme o processo.
No caso de um processo a pressão constante e de um processo a volume constante, teremos,
respectivamente, o calor específico a pressão constante (cp) ou o calor específico a volume
constante (cv).
Na tabela seguinte são dados valores do calor específico a pressão constante para algumas
substâncias.
Calor específico (J kg-1 K-1)
Sólidos elementares
Alumínio
900
Ferro
448
Cobre
387
Ouro
129
Chumbo
128
Outros sólidos
Madeira
1700
Vidro
837
Gelo (-5ºC)
2090
Mármore
860
Granito
670
Basalto
837
Líquidos
Água (15ºC)
4186
Mercúrio
140
Álcool (etílico)
2400
Substância
Exercício sugerido
Considerando o valor para a energia térmica recebida sob a forma de radiação solar na superfície
do globo terrestre de 180 Wm-2 (valor razoável para um local a cerca de 40ºN), calcule a taxa de
variação de temperatura de 1 kg de água e de 1 kg de granito. Que conclusão pode retirar dos
resultados?
Se em vez de nos referirmos à unidade de massa, nos referirmos a 1 mole da substância, então a
capacidade calorífica por mole designa-se por calor específico molar.
Calor latente
Quando se transfere energia térmica para uma substância, em geral a temperatura desta aumenta.
Há no entanto situações em que isto não acontece, como no caso em que a substância sofre uma
transição de fase (da fase sólida para a líquida: fusão; da fase líquida para a de vapor:
vaporização, etc.). Nestes casos, enquanto durar a transição de fase, a energia que está a ser
transferida para a substância é utilizada na mudança da sua estrutura molecular (o que envolve
variações da energia interna) e a substância mantém a temperatura.
A energia térmica necessária para mudar a fase de uma dada massa (m) de substância pura é dada
por:
Q=mL
26
TERMODINÂMICA APLICADA
2009 Isabel Ambar
em que L, o calor latente da substância, depende da natureza da mudança de fase e das
propriedades da substância (por exemplo, da temperatura, da pressão). A tabela seguinte mostra
os valores do calor latente de fusão e do calor latente de vaporização e as temperaturas a que se
dão as respectivas transições de fase (em condições de pressão normal), para diversas
substâncias. Para uma dada substância, o valor do calor latente de vaporização é muito maior
(uma ordem de grandeza ou mais) que o de fusão. Este facto não é de estranhar porque, para
contrariar as forças atractivas entre as moléculas de um líquido e conseguir separá-las tanto como
o que normalmente corresponde à fase de vapor, é necessária mais energia do que para passar de
uma estrutura sólida fortemente ordenada para uma estrutura líquida um pouco menos ordenada.
Fusão
Substância
Tra de fusão
(ºC)
- 209,97
- 218,79
0,00
327,3
660
1063,00
1083
Azoto
Oxigénio
Água
Chumbo
Alumínio
Ouro
Cobre
Vaporização
Calor latente (J kg-1)
2,55 x 104
1,38 x 104
3,33 x 105
2.45 x 104
3,97 x 105
6,44 x 104
1,34 x 105
Tra de vaporização
(ºC)
- 195,81
- 182,97
100,00
1750
2450
2660
1187
Calor latente (J kg-1)
2,01 x 105
2,13 x 105
2,26 x 106
8,70 x 105
1,14 x 107
1,58 x 106
5,06 x 106
Exercício sugerido
Considere a conversão de 1 kg de gelo (à temperatura de –5,0 ºC) em vapor de água a 120,0ºC.
Calcule a energia térmica necessária para esta conversão (utilize o valor de 2,01 x 103 J kg-1 K-1
para o calor específico do vapor entre 100 ºC e 120ºC).
T (ºC)
120
100
vapor
0
- 30
água
gelo gelo+água
água
+
vapor
Energia térmica fornecida
4.3. Transferência de energia sob a forma de trabalho
Podemos definir trabalho como uma transferência de energia entre um sistema e a sua vizinhança
cujo único efeito externo às fronteiras do sistema pudesse ter sido a elevação de um peso. O
trabalho realizado por (ou sobre) um sistema corresponde a uma transferência de energia entre o
sistema e a sua vizinhança. Não tem sentido falar de trabalho de um sistema mas sim de trabalho
realizado (ou recebido) pelo sistema, porque para haver trabalho tem de haver um processo de
transferência de energia sob essa forma. O trabalho do ponto de vista termodinâmico pode
27
TERMODINÂMICA APLICADA
2009 Isabel Ambar
coincidir com o trabalho mecânico (no caso de sistemas simples) mas pode também corresponder
a um trabalho químico, magnético, eléctrico, etc. O trabalho realizado pelos sistemas pode ser
expresso sob a forma do produto de uma variável intensiva pela variação infinitesimal da variável
extensiva conjugada. Por agora vamos considerar apenas o trabalho mecânico associado à
variação de volume do sistema, mas a tabela junta mostra a expressão do trabalho para vários
tipos de sistemas.
Sistema
Hidrostático
Fio extensível
Película superficial
Célula eléctrica
Sólido magnético
Variável intensiva
(força generalizada)
Variável extensiva
(deslocamento
generalizado)
Pressão, p
Força aplicada, F
Tensão superficial, σ
Força electromotriz, ε
Intensidade magnética,
H
Volume, V
Comprimento do fio, L
Área superficial, A
Carga eléctrica, Z
Magnetização, M
Trabalho
- p dV
F dL
σ dA
ε dZ
H dM
Trabalho mecânico
Considere-se um gás contido num cilindro com um êmbolo móvel. O gás ocupa um volume (V) e
exerce uma pressão (p) sobre as paredes e o êmbolo. Se este tiver uma secção recta com uma área
A, a força exercida pelo gás sobre o êmbolo será o produto da pressão pela área (p A).
Supondo que o gás se expande de um modo quase-estático (i.e., tão lentamente que o sistema se
mantém essencialmente em equilíbrio ao longo de todo o processo) deslocando o êmbolo de uma
distância infinitesimal dl, então o trabalho realizado será pAdl. Vamos adoptar a seguinte
convenção: o trabalho é negativo ou positivo conforme o sistema aumenta ou diminui de volume.
Isto corresponde ao facto de que quando o sistema aumenta (diminui) de volume ele está a
realizar (receber) trabalho e portanto a perder (ganhar) energia e daí o sinal negativo (positivo).
dl
Então o trabalho infinitesimal realizado pelo gás será:
đW = - p dV
Escrevemos đW e não dW (que corresponderia à diferencial de uma função W) porque o trabalho
(W) não corresponde a uma função de estado: o trabalho depende do processo que leva o sistema
28
TERMODINÂMICA APLICADA
2009 Isabel Ambar
do estado inicial ao estado final. Portanto, W não é diferenciável. đW significa apenas a
transferência de uma quantidade infinitesimal de energia sob a forma de trabalho.
Note-se que há dois requisitos para que se dê uma interacção do tipo trabalho mecânico entre um
sistema e a sua vizinhança: (i) tem de haver uma força a actuar sobre a fronteira do sistema e (ii)
a fronteira tem de se mover. O deslocamento da fronteira sem haver qualquer força que se oponha
ou que cause o movimento (tal como ocorre durante a expansão livre de um gás para uma região
de vácuo) não corresponde a um trabalho.
Num processo quase-estático existe praticamente equilíbrio mecânico em todas as etapas do
processo. Então num processo quase-estático finito, no qual o volume varia de V1 para V2, o
trabalho mecânico é dado pelo integral:
V2
W = − ∫ p(V ) dV
V1
em que p(V) significa que a pressão varia com o volume do sistema. Num gráfico que tenha
como coordenadas p e V (diagrama de Clapeyron), o trabalho corresponderá à área sob a curva
que representa a variação de p em função de V:
p
1
2
V1
V2
V
Na figura, o trabalho é negativo pela convenção acima referida (sistema aumenta de volume
realizando trabalho). Se o processo quase-estático que liga os estados 1 e 2 fosse representado por
uma curva diferente, a área que mede o trabalho realizado seria diferente. Então concluímos que
o trabalho depende não só dos estados inicial e final do sistema mas também do próprio processo
entre esses estados.
Se o processo for cíclico, i.e., se o estado inicial e final coincidirem, o trabalho pode ser diferente
de zero.
W = − ∫ p dV ≠ 0
A representação, num diagrama p-V, do trabalho resultante num processo cíclico corresponde à
área envolvida pela curva que representa o processo.
p
V
29
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Exercícios sugeridos
Calcule o trabalho mecânico realizado num processo quase-estático entre dois estados, nos seguintes
casos:
a) Processo isocórico (a volume constante).
b) Expansão isobárica (a pressão constante) entre os volumes V1 e V2.
c) Expansão isotérmica (a temperatura constante) de um gás ideal.
d) Expansão adiabática (sem trocas de calor) de um gás ideal, sabendo que a relação entre p e V
nesse processo é dada por pVγ = Cte, onde γ (>1) é o coeficiente adiabático do gás (γ = cp/cv).
4.4. Primeiro Princípio da Termodinâmica. Aplicações.
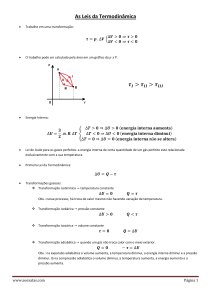
A primeira lei da Termodinâmica corresponde a uma lei de conservação da energia em que se
consideram as trocas de energia sob a forma de calor e de trabalho e a variação da energia
interna. Supondo então que um sistema sofre uma mudança de um estado (estado inicial) para
outro estado (estado final), durante a qual troca calor (Q, positivo se for transferido para o
sistema) e trabalho (W, positivo se for transferido para o sistema). A quantidade Q + W, se for
medida em diferentes processos que levem o sistema do mesmo estado inicial para o mesmo
estado final, é exactamente a mesma para todos esses processos. A esta quantidade, que é
completamente determinada pelos estados inicial e final, não dependendo da forma como o
sistema é levado de um para o outro estado, chamamos variação da energia interna do sistema
(ΔU = Uf – Ui). Então a expressão deste facto constitui o primeiro princípio da Termodinâmica:
ΔU = Uf – Ui = Q + W
Se um sistema sofrer uma variação infinitesimal de estado, em que quantidades muito pequenas
de trabalho (đW ) e de calor (đQ ) são trocadas com o exterior, a 1ª lei pode ser escrita na forma:
dU = đQ + đW
onde dU representa uma diferencial exacta, ou seja a diferencial de uma função de estado. A
variação de uma função de estado entre dois estados só depende destes. Repare-se que no caso do
trabalho e do calor utilizámos o d cortado (đ) justamente para assinalar que não se trata do
operador diferencial visto que nem Q nem W são funções de estado. Ao nível microscópico, a
energia interna de um sistema inclui a energia potencial e a energia cinética das moléculas que o
constituem. Considerando apenas sistemas simples em que o trabalho será apenas mecânico
(đW= - pdV), então para um processo quase-estático podemos escrever:
dU = đQ – p dV
Consideremos alguns casos particulares de processos com relevância:
(i) Sistema isolado (sem interacção com a vizinhança)
Como neste caso Q = W = 0, então:
ΔU = 0
A energia interna de um sistema isolado mantém-se constante.
30
TERMODINÂMICA APLICADA
2009 Isabel Ambar
(ii) Sistema não isolado, processo cíclico
Como o processo é cíclico, os estados inicial e final são iguais e portanto a variação de energia
interna (ΔU) é nula. Então:
Q+W=0
o que significa que a energia térmica fornecida ao sistema (Q > 0) é igual ao trabalho realizado
pelo sistema (W < 0), ou que a energia térmica fornecida pelo sistema (Q < 0) é igual ao trabalho
realizado sobre o sistema (W > 0).
(iii) Processo adiabático
Um processo adiabático caracteriza-se por não haver trocas de energia térmica entre o sistema e a
vizinhança, o que na prática se consegue isolando termicamente o sistema ou realizando muito
rapidamente o processo. Então Q = 0 e portanto:
ΔU = W
No caso, por exemplo, de um gás, se este se expandir adiabaticamente (W < 0, Q = 0), a energia
interna terá de decrescer (ΔU < 0) e a temperatura do gás diminuir. Se o gás for comprimido
adiabaticamente, a sua temperatura vai aumentar (um exemplo prático é o aquecimento de uma
bomba de encher os pneus da bicicleta, e que é devido ao aquecimento do ar ao ser comprimido
no interior da bomba).
(iv) Processo isocórico (volume constante)
Se considerarmos apenas o trabalho mecânico (-p dV), neste caso como o volume se mantém
constante, o trabalho será nulo e portanto a energia térmica trocada com a vizinhança é igual à
variação da energia interna do sistema:
ΔU = Q
Durante a explosão da mistura de vapor de gasolina e ar no cilindro de um motor (de explosão), a
temperatura e a pressão sobem bruscamente porque o volume do cilindro não varia quase nada
durante a curta duração da explosão.
(v) Expansão livre adiabática de um gás
Considerando um gás que se expande livremente para uma região de vácuo isolada termicamente,
não há trabalho posto em jogo (o gás não realiza nem recebe trabalho) e portanto Q = W = 0.
Então neste processo será:
ΔU = 0
Os valores da energia interna inicial e final de um gás coincidem numa expansão livre adiabática.
No caso de um gás ideal, como a energia interna depende apenas da temperatura (como vimos),
não haverá variações de temperatura do gás (ideal) numa expansão livre adiabática.
A partir do 1º princípio ou lei da Termodinâmica vemos que se pode aumentar a energia de um
sistema, quer realizando trabalho sobre este quer fornecendo-lhe calor. O resultado final é o
mesmo mas a escolha de um ou outro modo de transferência de energia depende apenas das
conveniências (para aquecer água não dá muito jeito agitar pás no seu interior mas sim levar o
recipiente ao lume...). No caso da transferência de energia se fazer sob a forma de trabalho, este
pode ser sempre quantificado em termos mecânicos (relacionando-se com o deslocamento de um
peso), mas no caso do calor não. Há um outro aspecto importante que distingue calor e trabalho e
que será visto mais detalhadamente quando apresentarmos a 2ª lei da Termodinâmica: todo o
31
TERMODINÂMICA APLICADA
2009 Isabel Ambar
trabalho realizado sobre um sistema se pode transformar, através de um processo cíclico, em
calor, mas nem todo o calor absorvido se pode transformar ciclicamente em trabalho.
Uma máquina térmica é um dispositivo que trabalha por ciclos (ao fim de cada ciclo, a energia
interna do sistema volta ao valor inicial) e que produz trabalho à custa de calor que lhe é
fornecido. Uma máquina que produzisse trabalho sem dispêndio de calor – Máquina de
Movimento Perpétuo de Primeira Espécie - seria o ideal mas não é possível porque viola o 2º
princípio da Termodinâmica, como veremos.
5. Máquinas térmicas e frigoríficas. Segundo Princípio da Termodinâmica. Processos
reversíveis e irreversíveis. Máquina de Carnot.
A 1ª lei da Termodinâmica corresponde a um princípio de conservação de energia generalizado,
no qual se inclui o calor como uma das formas de transferência de energia. Mas este princípio
não estabelece quaisquer restrições relativamente às transferências de energia que podem ocorrer,
não fazendo qualquer distinção entre trabalho e calor. Mas há de facto diferenças entre estes dois
tipos de transferência de energia e é isso que a 2ª lei da Termodinâmica estabelece. Há processos
que são consistentes com a 1ª lei mas que não ocorrem naturalmente. Por exemplo, quando dois
objectos a temperaturas diferentes são postos em contacto térmico há transferência de calor do
que está a maior temperatura para o outro, mas a transferência em sentido contrário não ocorre
espontaneamente. Este procedimento está de acordo com a 2ª lei da Termodinâmica.
5.1. Máquinas térmicas e frigoríficas.
5.1.1. Máquinas térmicas
Uma máquina térmica é um dispositivo que converte energia térmica em outras formas úteis de
energia, tal como a energia mecânica ou a energia eléctrica. Por exemplo, na produção de
electricidade numa central térmica, utiliza-se carvão ou outro combustível para se produzir
energia térmica que, por sua vez, converte água em vapor, o qual vai accionar as pás de uma
turbina pondo-a em rotação. Esta energia mecânica é então utilizada num gerador eléctrico. Outro
tipo de máquina térmica é o motor de combustão interna do automóvel, que extrai energia
térmica da combustão da gasolina e converte parte dela em energia mecânica.
Numa máquina térmica há uma substância que sofre um processo cíclico durante o qual o
dispositivo recebe energia térmica (Qq) de uma fonte de calor a alta temperatura (Tq), a fonte
quente, realiza trabalho (W) e cede energia térmica (Qf ) a uma fonte de calor a baixa temperatura
(Tf), a fonte fria. Como a substância sofre um processo cíclico, a variação da sua energia interna é
nula e portanto, pela 1ª lei, virá:
Q + W = 0 ou (Qq +Qf) + W = 0 ou, exprimindo em módulos,
(⎜Qq⎜ - ⎜Qf⎜) - ⎜W⎜ = 0
onde W é < 0 visto ser trabalho realizado pelo sistema.
Tq
Qq
W
Qf
Tf
32
TERMODINÂMICA APLICADA
2009 Isabel Ambar
O rendimento de um dispositivo é sempre uma razão entre o que se pretende obter com esse
dispositivo e aquilo que se tem de “pagar”. No caso duma máquina térmica, o que se pretende é
obter trabalho (W) e o que se tem de “pagar” é a energia térmica que o dispositivo vai buscar à
fonte quente. Então o rendimento de uma máquina térmica é:
η=
W
Qq
Uma máquina térmica com rendimento de 100% teria de converter em trabalho toda a energia
térmica absorvida e isso a experiência diz-nos que não é possível. Só uma fracção dessa energia
térmica é que é convertida (por exemplo, um motor de automóvel tem um rendimento de cerca de
20% se for a gasolina e entre 35% e 40% se for a diesel). Com base nestes factos, a 2ª lei da
Termodinâmica pode ser enunciada na seguinte forma - enunciado de Kelvin-Planck:
É impossível uma máquina térmica que, operando por ciclos, não produza outro efeito
para além de absorver uma dada quantidade de energia térmica de um reservatório e
produzir uma quantidade igual de trabalho.
A transformação completa de energia térmica em trabalho seria possível mas nunca num processo
cíclico. Uma máquina térmica que operando por ciclos transformasse completamente em trabalho
uma certa quantidade de calor extraída de uma fonte (isto é, trabalhasse apenas com uma fonte
quente) seria uma Máquina de Movimento Perpétuo de Segunda Espécie.
5.1.2. Máquinas frigoríficas e bombas térmicas
Uma máquina frigorífica e uma bomba térmica ou bomba de calor correspondem a máquinas
térmicas a trabalhar em sentido inverso: a máquina absorve energia térmica (Qf) da fonte fria e
cede energia térmica (Qq) à fonte quente. Claro que isto só se pode realizar se a máquina receber
trabalho (W > 0). Neste caso, a 1ª lei aplicada ao sistema será:
Q+W=0
ou (Qf + Qq) + W = 0 ou, exprimindo em módulos, ⎜Qf ⎜– ⎜Qq⎜ + ⎜W⎜ = 0
onde W é >0 visto ser trabalho recebido pelo sistema.
Um frigorífico transfere energia térmica de um corpo mais frio (o interior do frigorífico) para um
corpo mais quente (a sala onde está) e isso faz-se à custa de trabalho que lhe é fornecido (e que
aparece na conta da electricidade ao fim do mês...). Se não fosse preciso fornecer trabalho
teríamos um frigorífico perfeito, o que seria mais uma violação da 2ª lei da Termodinâmica, a
qual pelo enunciado de Clausius tem a forma seguinte:
É impossível uma máquina que, operando por ciclos, não produza outro efeito senão
transferir energia térmica de um corpo para outro a temperatura mais alta.
Quer dizer, a energia térmica não flui espontaneamente de um objecto frio para um objecto
quente.
Vejamos qual é a diferença entre uma máquina frigorífica e uma bomba térmica, se ambos os
dispositivos funcionam do mesmo modo. Em ambos, temos de fornecer trabalho (W), mas a
diferença está no que se pretende obter com o desempenho do dispositivo: no caso do frigorífico
o objectivo é arrefecer um corpo (i.e., retirar Qf) enquanto que, no caso da bomba térmica, o que
33
TERMODINÂMICA APLICADA
2009 Isabel Ambar
se pretende é aquecer um corpo (i.e., fornecer Qq). Então, por definição, os coeficientes de
eficiência (rendimentos) desses dois dispositivos terão expressões diferentes:
Tq
Qq
Tq
Qq
W
W
Qf
Tf
Qf
Tf
Máquina frigorífica: coeficiente de eficiência
η=
Bomba térmica: coeficiente de eficiência
Qf
η=
W
Qq
W
Nota: Ao utilizar as expressões para o cálculo dos rendimentos ou dos coeficientes de eficiência,
utilizar os valores absolutos das grandezas Qq, Qf e W.
5.2. Processos reversíveis e irreversíveis. Máquina de Carnot.
Antes de apresentar a máquina térmica com o maior rendimento possível, temos de ver qual a
diferença essencial entre reversibilidade e irreversibilidade. Se no final da realização de um
processo reversível, este for invertido, tanto o sistema como a sua vizinhança voltam exactamente
ao estado inicial, sem haver quaisquer modificações do resto do universo. Um processo que não
satisfaça estas condições é um processo irreversível. Uma característica dos processos reversíveis
é não haver efeitos dissipativos, tais como os associados ao atrito, à turbulência, à resistência
eléctrica, à histerese magnética, etc., que convertem energia mecânica, eléctrica ou magnética,
em energia térmica. Na realidade, esses efeitos são impossíveis de eliminar totalmente e portanto
todos os processos naturais são irreversíveis. No entanto, alguns são quase-reversíveis.
É possível, em laboratório, aproximar as condições de realização de um processo daquelas
exigidas para ser um processo reversível. Por exemplo, se se deixar um gás no interior de um
cilindro (provido de um êmbolo bem lubrificado para evitar atritos nas paredes) expandir-se
muito lentamente, esse gás sofre um processo aproximadamente reversível.
Carnot, em 1824, descreveu uma máquina térmica ideal como tendo um rendimento máximo
possível ao operar num ciclo reversível – ciclo de Carnot – entre dois reservatórios de calor
(teorema de Carnot). Quer dizer que o trabalho resultante de uma máquina de Carnot é o máximo
possível para uma dada quantidade de calor fornecida pelo reservatório à temperatura mais alta.
Vejamos os passos principais de um ciclo de Carnot supondo que a substância consiste num gás
ideal contido num cilindro com um êmbolo móvel (tanto o cilindro como o êmbolo são
adiabáticos):
(i) A→B: Expansão isotérmica do gás, em contacto térmico com um reservatório à
temperatura Tq; o gás absorve a quantidade de calor Qq e realiza um trabalho WAB ao
empurrar o êmbolo;
34
TERMODINÂMICA APLICADA
2009 Isabel Ambar
(ii) B→C: Expansão adiabática do gás; a base do cilindro é substituída por um material não
condutor e o gás expande-se adiabaticamente; a temperatura do gás baixa de Tq para Tf
enquanto o gás realiza trabalho, WBC.
(iii)C→D: Compressão isotérmica do gás, em contacto térmico com um reservatório à
temperatura Tf < Tq; o gás expele a quantidade de calor Qf para o reservatório e recebe um
trabalho WCD de um agente externo;
(iv) D→A: Compressão adiabática do gás; a base do cilindro é substituída novamente por um
material não condutor e a temperatura do gás aumenta até Tq; o gás recebe um trabalho
WDA de um agente externo. Numa compressão adiabática a pressão sobe mais bruscamente
(pVγ=cte.) do que numa compressão isotérmica (pV=cte.) porque a própria subida da
temperatura intensifica a subida da pressão.
O trabalho total realizado neste processo cíclico reversível corresponde, num diagrama pV, à área
circundada pelas curvas representativas do processo ABCDA. Como a variação de energia
interna do gás num ciclo é nula, o trabalho resultante tem de ser igual à quantidade de energia
térmica transferida para o sistema: Wres + (⎜Qq⎜ – ⎜Qf⎜) = 0 ou Wres = (⎜Qf ⎜– ⎜Qq⎜)<0. O módulo
do trabalho resultante é |Wres| = (⎜Qq⎜ – ⎜Qf⎜) e o rendimento desta máquina é:
η carnot =
Mas
Wres
Qq
=
Qq − Qf
= 1−
Qq
Qf
Qq
Qf
T
T
= f (ver exercício sugerido) e portanto: η carnot = 1 − f
Tq
Tq
Qq
Qq
A
p
B
A
Qq
B
W
D
B
A
C
D
Qf
C
V
Qf
C
D
35
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Podemos então afirmar que: todas as máquinas de Carnot operando reversivelmente entre as
mesmas duas temperaturas têm o mesmo rendimento. E esse rendimento é maior do que o de
qualquer máquina irreversível (real) operando entre essas mesmas temperaturas. O rendimento
máximo é, no entanto, sempre menor que 100% (η = 100% corresponderia a Tf = 0 K).
O trabalho reversível (máximo) será dado pela expressão:
Qq
T
Wrev = η carnot Q q = (1 − f ) Q q = (Tq − Tf )
Tq
Tq
Exercício sugerido
Mostre que o rendimento de uma máquina de Carnot utilizando um gás ideal pode ser dado pela
T
expressão η = 1 − f
Tq
Sugestão:
(i) Calcule as quantidades Qq e Qf durante as fases de expansão e compressão isotérmica
V
ln( C
)
Q
T
VD .
(respectivamente, A→B e C→D) e mostre que f = f
Q q Tq ln( VB
)
VA
Note que na expansão isotérmica (A→B) de um gás ideal, a energia interna não varia (U é função
exclusiva de T para um gás ideal) e portanto é nula a soma do calor trocado com a fonte quente
(Qf) com o trabalho realizado pelo sistema E o mesmo se dá na compressão isotérmica (C→D).
VB
))>0
VA
V
Qf = - WCD = - (- nRuTf ln( D ))<0
VC
Qq = - WAB = - (- nRuTq ln(
Então:
Qf
Qq
=
WCD
− W AB
(ii) Sabendo que num processo adiabático de um gás ideal as seguintes relações são válidas:
pVγ= cte. e TVγ−1 = cte. (γ é a razão entre os calores específicos a pressão e a volume constante),
aplique a relação relevante nos processos adiabáticos B→C e D→A, de modo a obter:
VB VC
=
V A VD
Vejamos agora o funcionamento de uma máquina frigorífica, cujo objectivo, como vimos, é
manter um corpo a baixas temperaturas (o qual vai ser identificado como a fonte fria) retirandolhe uma certa quantidade de calor em cada ciclo. Para que seja possível transferir calor de uma
fonte fria para uma fonte quente (a atmosfera ambiente, por exemplo), é preciso fornecer trabalho
à máquina. Vamos então considerar o ciclo de Carnot percorrido em sentido contrário ao da
máquina térmica.
36
TERMODINÂMICA APLICADA
2009 Isabel Ambar
p
D
Qq
C
W
A
Qf
B
V
Ciclo de Carnot para uma máquina frigorífica:
(i) A→B: vaporização do fluido que está na fase líquida em A (T=Tf constante) no
evaporador; o vapor é aspirado no cilindro do compressor pelo movimento do êmbolo e,
durante esta operação, o fluido retira Qf da fonte fria;
(ii) B→C: compressão adiabática do vapor no cilindro, com aumento da temperatura (Tf
→Tq);
(iii) C→D: condensação do vapor na serpentina arrefecida pela circulação de água; o fluido
cede calor (Qq) à fonte quente;
(iv) D→A: expansão adiabática do líquido, com abaixamento da temperatura (Tq → Tf). Na
realidade, este processo não é adiabático reversível mas sim isentálpico (como veremos
mais adiante): o fluido é obrigado a atravessar uma barreira porosa de uma região de alta
pressão para uma de baixa pressão e arrefece.
Líquido a
alta pressão
p
Mistura líq.+vapor
a baixa pressão e T
Transferência de calor para o
meio ambiente
Vapor a
alta pressão
Condensador
(condensador)
(traseiraQdo
frigoríf.)
q
D
C
Compressor
Válvula de expansão
(traseira do
(tubo capilar longo) W
frigoríf, em baixo)
A
B
Qf
(evaporador)
Evaporador
(em torno do congelador, no
interior do frigoríf.)
V
Trabalho
Vapor a
baixa pressão
Transferência de calor do espaço
refrigerado
Fluido refrigerante normalmente utilizado nos frigoríficos: amoníaco, gás carbónico, fréons (ou
clorofluorcarbonetos, cujo uso é actualmente proibido por causa do efeito destrutivo sobre a
37
TERMODINÂMICA APLICADA
2009 Isabel Ambar
camada de ozono). Este fluido circula entre o evaporador (sistema de tubos em serpentina) e o
condensador (sistema de tubos em serpentina na parte exterior do frigorífico, na traseira) por meio
de um compressor (que é activado por um motor). O refrigerante líquido sai do condensador e
segue para o evaporador (no interior do frigorífico; na parede do congelador) onde o ar dentro do
frigorífico é arrefecido por contacto com os tubos onde circula o fluido refrigerante.
Um frigorífico doméstico mantém uma temperatura no congelador de cerca de -18ºC e o
compartimento refrigerado entre 2º e 8ºC. A gaveta dos vegetais e fruta fresca tem como objectivo
proteger estes do efeito de secura do ar frio (ar frio tem pouca capacidade de retenção de vapor de
água) que circula no interior do frigorífico.
Condensador de um frigorífico
No caso de uma bomba térmica, o esquema de funcionamento é semelhante ao da máquina
frigorífica, mas agora o objectivo é utilizar a quantidade de calor cedida à fonte quente para
aquecimento. A fonte fria pode ser, por exemplo, a atmosfera exterior a uma sala ou a água de
uma ribeira, e a fonte quente o radiador (convector) de água quente ou um evaporador industrial.
As expressões do coeficiente de eficiência de uma máquina frigorífica reversível (ηfrig) e do
coeficiente de eficiência de uma bomba térmica reversível (ηbterm) podem também ser dadas em
função das temperaturas:
Qq
Tq
Qf
Tf
η frig =
=
η bterm =
=
W
Tq − Tf
W
Tq − Tf
Note-se que ηfrig>1 ou <1 e ηbterm>1.
Exemplo:
Considere-se a instalação de uma “chauffage” por radiadores (com temperatura de 50ºC) sendo a
fonte fria a atmosfera exterior a 12ºC. Supondo a máquina reversível, o coeficiente de eficiência
323
será: η bterm =
= 8,5 . Se o compressor gastar 1kWh, a fonte quente receberá uma quantidade
38
de calor Qq= ηbterm W = 8,5 x 3600 kJ = 30.600 kJ. A transformação directa daquele trabalho em
38
TERMODINÂMICA APLICADA
2009 Isabel Ambar
calor utilizando, por exemplo, um radiador eléctrico (efeito Joule), daria no máximo uma
quantidade de calor de 3.600 kJ.
É preciso notar, no entanto, que o coeficiente de eficiência real de uma bomba térmica tem,
geralmente, valores entre 2 e 3, porque há imensas causas de irreversibilidade (tais como o atrito)
que tornam o processo menos rentável do que no caso reversível.
5.3. Escala absoluta de temperaturas
Vimos que as escalas de temperatura que foram definidas (ponto 2.2) utilizavam a variação de
propriedades físicas de algumas substâncias com a variação da temperatura. Mas seria importante
que se definisse uma escala que fosse independente das propriedades dos materiais. Uma
máquina de Carnot oferece essa possibilidade visto que:
- todas as máquinas térmicas reversíveis (máquinas de Carnot) têm o mesmo rendimento quando
operam entre as mesmas duas temperaturas (i.e., o rendimento é independente do fluido que
está a operar e das suas propriedades) e o seu rendimento é função exclusiva dessas
temperaturas.
η carnot = 1 −
Qf
Qq
= 1−
Tf
Tq
Então, a razão Qf/Qq depende apenas das temperaturas dos dois reservatórios e portanto a razão
Tf/Tq pode ser obtida operando uma máquina térmica reversível num ciclo de Carnot entre essas
duas temperaturas e medindo cuidadosamente Qf e Qq. Na escala de temperaturas absolutas de
Kelvin, as razões das temperaturas dependem apenas das razões entre as quantidades de calor
transferidas entre uma máquina térmica reversível e os dois reservatórios. Mas isto apenas nos dá
uma razão entre temperaturas absolutas. Podemos, no entanto, obter uma escala de temperaturas
relativa a alguns pontos fixos. A escala absoluta de temperaturas é definida escolhendo o valor
de 273,16 K como correspondendo ao ponto triplo da água. Um grau Kelvin é definido como
1/273,16 do intervalo de temperaturas entre o zero absoluto e o ponto triplo da água. Então a
temperatura de qualquer substância poderia ser obtida levando-a a percorrer um ciclo de Carnot e
medindo a energia térmica Q recebida (ou cedida) pelo sistema à temperatura desconhecida T e a
energia térmica Qtriplo cedida (ou recebida) pelo sistema à temperatura do ponto triplo da água:
Q
T = (273,16)
Q triplo
Claro que, do ponto de vista prático, este processo de medição de temperaturas não é possível e
há, como vimos, outros meios de o fazer (parágrafo 2.2).
6. Entropia. Variações de entropia em processos reversíveis e irreversíveis.
Assim como o conceito de temperatura foi introduzido a propósito da lei zero da Termodinâmica
e o conceito de energia interna no contexto da primeira lei, vamos agora, a propósito da segunda
lei, introduzir mais uma função de estado – a entropia (S). Foi Clausius (1865) quem introduziu
esta função de um ponto de vista macroscópico.
39
TERMODINÂMICA APLICADA
2009 Isabel Ambar
6.1. Desigualdade de Clausius
Considere-se um sistema S que sofre uma transformação cíclica. Durante o ciclo, o sistema troca
calor (Q1, Q2, ..., Qn) com um conjunto de n fontes a temperaturas T1, T2, ..., Tn. Vamos
demonstrar a desigualdade de Clausius:
n
Qi
≤0
Ti
i =1
∑
ciclo
onde o sinal de igual corresponde a um ciclo reversível.
A desigualdade de Clausius estabelece o seguinte:
Numa transformação cíclica, a soma dos calores recebidos e cedidos pelo sistema (com
os sinais positivos ou negativos, respectivamente) divididos pelas temperaturas
absolutas das fontes de calor, é negativa ou nula.
A soma é negativa se os processos forem irreversíveis e é nula se os processos forem reversíveis.
S
Qn
Q1
T1
Q2
...
T2
Tn
Demonstração
Para além das n fontes, vamos introduzir uma fonte de calor à temperatura To e n máquinas
reversíveis (n ciclos de Carnot C1, C2, ..., Cn) que trabalham entre as fontes T1, T2, ..., Tn e a fonte
To.
De modo geral, o ciclo de Carnot Ci vai operar entre Ti e To de modo que fornece à fonte Ti uma
quantidade de calor Qi igual à que é retirada a essa mesma fonte Ti pelo sistema S.
S
Qn
Q1
T1
Q1
Q2
...
T2
Tn
Qn
Q2
W1
W2
Q1,o
Q2,o
To
40
Wn
Qn,o
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Como já vimos, para uma máquina de Carnot, a razão entre Qq e Qf é igual à razão entre as
temperaturas Tq e Tf. Então podemos escrever:
Q1,o
Q1
=
To
T1
Qi, o
...
Qi
=
To
Ti
Considerando um ciclo do sistema S e um ciclo de cada uma das máquinas de Carnot, a
resultante das trocas de calor em cada uma das fontes T1, T2, ..., Tn é nula. A fonte To perde uma
quantidade de calor Qo que é igual à soma das quantidades de calor absorvidas pelos ciclos C1,
C2, ..., Cn:
n
n
Qi
1
1 Ti
O resultado de todo este ciclo complexo (em que o sistema S e as máquinas de Carnot voltam aos
respectivos estados iniciais) vai corresponder ao conjunto (S+máquinas de Carnot) receber Qo da
fonte a temperatura uniforme To e transformá-lo inteiramente em trabalho.
Qo = ∑ Qi,o = To ∑
Sistema S + máq.
Carnot
W = W1 + W2 + …
Qo
To
Se Qo fosse positivo (i.e., se o sistema complexo recebesse calor) isto contrariava o enunciado de
Kelvin do 2º princípio da Termodinâmica. Então terá de ser:
n
Q
(6.1.1)
Qo ≤ 0
∑ Ti ≤ 0
i
1
Se o ciclo realizado por S for reversível, podemos invertê-lo e todos os Qi mudam de sinal. Então,
aplicando aquela desigualdade a este caso, virá:
n
n
- Qi
Q
≤
0
ou
(6.1.2)
∑T
∑ Ti ≥ 0
i
i
1
1
Então, no caso reversível, a compatibilização das expressões (6.1.1) e (6.1.2) implica:
n
Qi
=0
Ti
i =1
∑
ciclo
No caso de o sistema trocar calor com uma distribuição contínua de fontes, as somas devem ser
substituídas por integrais abrangendo o ciclo completo:
41
TERMODINÂMICA APLICADA
2009 Isabel Ambar
∫
δQ
T
≤0
onde o sinal de igual corresponde a um ciclo reversível.
T é a temperatura da fonte que troca calor com o sistema mas não é, necessariamente, igual à
temperatura T’ do sistema (ou de parte deste) que recebe o calor δQ.
Se o ciclo for irreversível, T’ ≤ T quando δQ for positivo (porque o calor não pode fluir de um
corpo mais frio para um mais quente) e T’ ≥ T se δQ for negativo.
Se o ciclo for reversível, teremos de ter sempre T’=T porque uma troca de calor entre dois corpos
a temperaturas diferentes não é reversível. Então T neste caso tem de ser a temperatura da fonte e
também a temperatura da parte do sistema que recebe o calor δQ.
6.2. A entropia
∫
δQ
= 0 no caso de um ciclo reversível. Dado que o integral cíclico de uma grandeza
T
que depende apenas do estado do sistema (i.e., uma propriedade do sistema), é nulo, então a
δQ
grandeza
deverá representar uma propriedade. Clausius, em 1865, descobriu essa
T
propriedade e denominou-a entropia (para a qual se utiliza a letra S). A entropia é uma
propriedade extensiva e tem unidades de energia/temperatura (no sistema SI, será J/K).
Vimos que
Sejam A e B dois estados de equilíbrio de um sistema S e considerem-se diferentes processos
B
reversíveis entre estes dois estados. Vamos mostrar que
δQ
∫T
é o mesmo para todos os processos
A
reversíveis entre A e B, e portanto, que o valor do integral numa transformação reversível só
depende dos estados inicial e final e não da transformação.
(i)
•B
Considerem-se então dois processos reversíveis, (i) e (ii), de A para B.
Como o ciclo A→(i)→B→(ii)→A é reversível (é composto por dois
A•
(ii)
processos reversíveis), então:
δQ
∫ T =0
AiBiiA
Mas este integral pode ser subdividido em duas partes:
⎡A δQ ⎤
δQ ⎤
⎥ =0
⎢∫
⎥ + ⎢∫
⎣⎢A T ⎥⎦ i ⎢⎣ B T ⎦⎥ ii
1424
3
⎡B
⎤
δQ ⎥
− ⎢⎢ ∫
⎥
⎢⎣ A T ⎥⎦
ii
⎡B
⎡ B δQ ⎤ ⎡ B δQ ⎤
⎢∫ ⎥ = ⎢∫ ⎥
⎣⎢A T ⎦⎥ i ⎢⎣A T ⎦⎥ ii
ou
Vamos escolher um certo estado de equilíbrio do sistema – estado O – como um estado de
referência. Se A for outro estado de equilíbrio, então:
42
TERMODINÂMICA APLICADA
2009 Isabel Ambar
⎡A δQ ⎤
⎢∫
⎥ =
⎢ T ⎥
⎣O ⎦ rev
S(A) = entropia do estado A
Considerando agora dois estados de equilíbrio A e B, sejam S(A) e S(B) as entropias desses
estados. Vamos mostrar que:
B
S(B) – S(A) =
∫
A
( rev)
B
Como
∫
A
( rev)
δQ
δQ
T
tem o mesmo valor para qualquer transformação reversível entre A e B, vamos
T
escolher uma transformação consistindo em dois processos reversíveis sucessivos: de A para o
estado de referência O, e deste para B:
B
∫
A
(rev)
δQ
=
T
O
∫
A
(rev)
δQ
+
T
1
424
3
A
δQ
− ∫
T
O
B
∫
O
(rev)
δQ
= S(B) − S(A)
T
1
424
3
S(B)
(rev)
14
24
3
−S(A)
A definição de entropia requer uma escolha arbitrária de um estado de referência. Se tivéssemos
escolhido outro estado de referência (O’), o novo valor da entropia do estado A diferia do antigo
(referido a O) apenas numa constante aditiva.
A
S’(A) =
∫
O'
rev
δQ
T
O
=
δQ
∫T
O'
A
+
δQ
∫T
O'
A
=+
δQ
O'
δQ
∫T ∫T
O'
−
= S(A) – S(O’)
O
S(A) – S’(A) = S(O’) = constante (independente do estado A).
A entropia é definida a menos de uma constante aditiva. Para calcular diferenças de entropia não
há necessidade de se conhecer essa constante aditiva; para a determinar teremos de aplicar a 3ª lei
da Termodinâmica (ponto 6.5).
δQ
só nos dá o valor da variação da entropia se a integração for levada
Note-se que o integral de
T
δQ
a cabo ao longo de um caminho reversível entre os dois estados. O integral de
ao longo de
T
um processo irreversível não corresponde à variação de uma propriedade e, em geral, pode tomar
valores diferentes conforme o caminho entre os estados inicial e final.
43
TERMODINÂMICA APLICADA
2009 Isabel Ambar
6.3. Variação de entropia num processo reversível
Considere-se um processo infinitesimal de um sistema entre dois estados de equilíbrio. Se δQr for
a quantidade de energia térmica transferida no caso de o sistema seguir um caminho reversível,
então a variação de entropia dS, independentemente do caminho seguido, é dada por:
dS =
δQ r
T
Quando o sistema absorve (rejeita) energia térmica δQ > 0 (δQ < 0) a entropia aumenta
(diminui).
Num processo reversível entre dois estados (inicial e final), a variação de entropia é dada por:
f
f
δQ
ΔS = ∫ dS = ∫ r
T
i
i
Como a entropia é uma função de estado, a variação da entropia de um sistema entre dois estados
é a mesma independentemente do caminho entre eles. Se o processo for adiabático (δQ = 0) e
reversível (δQr = T dS), então a variação de entropia é nula: o processo é isentrópico (dS = 0).
Se um sistema percorrer um ciclo reversível arbitrário, como a entropia é uma função de estado, a
f
δQ
variação ∆S = 0. Como ΔS = ∫ r então a variação de S no caso de um processo cíclico:
T
i
ΔS =
∫
δQ r
= 0
T
Supondo dois corpos que interagem reversivelmente, o que implica estarem em equilíbrio térmico
um com o outro (i.e., têm igual temperatura), e que uma pequena quantidade de energia térmica é
transferida de um para o outro corpo, então as correspondentes variações de entropia são (-δQ/T)
e (+δQ/T). A variação total de entropia do sistema formado pelos dois corpos é nula e portanto a
entropia do Universo mantém-se. A entropia do Universo não varia quando se dá um processo
reversível.
6.4. Variação de entropia num processo irreversível
Considere-se um processo cíclico que vai de A para B por um caminho reversível, e de B para A
por um caminho irreversível. A variação de entropia do sistema ao fim do ciclo é nula porque a
entropia é uma função de estado, e portanto a sua variação depende apenas desses estados e não
dos processos que ocorrem entre eles.
rev
∆Sciclo = ∆SAB + ∆SBA = 0
•B
Por outro lado, o teorema de Clausius diz-nos que:
B
Qirrev ,i
δQrev
+
∑
∫A T i Ti < 0
B→ A
44
A•
irrev
TERMODINÂMICA APLICADA
2009 Isabel Ambar
B
Mas como no processo reversível ΔS AB = ∫
δQrev
T
A
ΔS AB +
∑
, então esta desigualdade pode tomar a forma:
Qirrev,i
Ti
i
B→ A
<0
Como ∆SAB + ∆SBA = 0, então ∆SAB = -∆SBA e portanto podemos escrever:
Qirrev,i
− ΔS BA < − ∑
Ti
i
B→ A
Multiplicando a desigualdade por –1, vem:
Q
Q
[ΔSBA ]irrev > ∑ irrev,i ou, como os índices A e B são arbitrários, [ΔSAB ]irrev > ∑ irrev,i
Ti
Ti
i
i
B→A
A →B
Quer dizer que quando o sistema evolui de um estado para outro por contacto com diferentes
fontes, num processo irreversível, a variação de entropia é maior que a soma dos calores
cedidos ou recebidos pelo sistema divididos pelas temperaturas das fontes. Se o processo
irreversível se der em contacto com um número infinito de fontes:
B δQ
irrev
ΔS AB
> ∫
irrev
T
A
Ou, na forma infinitesimal:
δQ irrev
dS irrev >
T
A desigualdade, válida para os processos irreversíveis, mostra que há geração de entropia nesses
processos, a qual é devida inteiramente à presença das irreversibilidades:
B δQ
irrev + S
= ∫
ΔSAB
gerada
irrev
T
1
424
3
A
14243
>0
[
[
]
]
transfer.entropia
Se tivermos um processo irreversível que liga dois estados de equilíbrio, e quisermos determinar
a respectiva variação de entropia, basta considerar um processo reversível entre esses dois
estados e calcular ∫
δQ r
para esse processo. Mas aqui é preciso atenção, porque a quantidade de
T
energia térmica transferida de facto no processo irreversível (Qirrev) não coincide com a
quantidade Qr que seria transferida num processo reversível.
A entropia é uma propriedade extensiva: a entropia total de um sistema é igual à soma das
entropias das partes em que o sistema se subdivide.
Um sistema e a sua vizinhança constituem um sistema isolado visto que podem ser envolvidos
por uma parede através da qual não há transferências de calor, trabalho ou massa. Então a
transferência de entropia é nula e portanto a variação de entropia do sistema isolado é igual à
geração de entropia. Então, a variação da entropia desse conjunto (sistema+vizinhança) durante
um processo real será:
45
TERMODINÂMICA APLICADA
2009 Isabel Ambar
ΔStotal = ΔSsist + ΔSviz > 0
Como nenhum processo real é verdadeiramente reversível, podemos concluir que há sempre
geração de alguma entropia e portanto a entropia do universo (que é um sistema isolado) está
sempre a aumentar. Quanto mais irreversível for o processo, maior será a entropia gerada.
⎧> 0 processo irreversível
⎪
Sgerada ⎨= 0 processo reversível
⎪< 0 processo impossível
⎩
A segunda lei da Termodinâmica pode ser enunciada do seguinte modo:
A entropia total de um sistema isolado que sofre uma variação não pode decrescer.
O princípio da não diminuição da entropia corresponde a uma lei de evolução, dado que indica
em que sentido um sistema adiabático evolui. Na natureza, há uma tendência para a mudança até
ser atingido um estado de equilíbrio. Aquele princípio obriga a que a entropia de um sistema
isolado aumente até atingir um valor máximo compatível com as ligações a que o sistema está
sujeito. Nesse ponto, diz-se que o sistema atingiu o estado de equilíbrio visto que o princípio do
aumento de entropia impede o sistema de sofrer qualquer mudança de estado que resulte num
decréscimo de entropia. Então o estado de entropia máxima é o estado mais estável de um
sistema isolado.
Vimos então que, se o processo for irreversível a entropia total de um sistema isolado aumenta, se
for reversível a entropia total mantém-se constante. Portanto:
ΔSisolado ≥ 0
Mas será que não pode haver decréscimo de entropia de um sistema num processo irreversível?
Claro que pode, desde que esse decréscimo seja acompanhado por um aumento (maior em valor
absoluto) da entropia da vizinhança do sistema, de tal modo que a entropia do Universo (que é
um sistema isolado) não decresça. Isto é, a variação de entropia de um sistema pode ser negativa
durante um processo mas a geração de entropia é sempre positiva.
No caso do processo ser reversível, o decréscimo de entropia de um sistema terá de ser
compensado exactamente por um aumento (igual, em valor absoluto, àquele decréscimo) da
entropia da vizinhança, de modo que a entropia do Universo se mantenha constante.
O facto de todos os processos espontâneos de um sistema isolado serem tais que tendem a
aumentar a entropia do sistema pode ser ilustrado com os dois exemplos seguintes.
(i) Considere-se a troca de calor por condução térmica entre duas partes de um sistema,
respectivamente, à temperatura T1 e T2 (>T1). A variação da entropia da parte mais quente (a qual
vai perder calor) é (-Q/T2) e a da parte mais fria é (Q/T1). A variação total de entropia do sistema
é:
Q Q
−
>0
T1 T2
46
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Então a entropia total do sistema aumentou.
(ii) Considerando o processo (irreversível) da produção de calor por atrito, a parte do sistema que
aquece devido ao atrito recebe uma quantidade de calor positiva e a sua entropia aumenta. Como
o calor proveio do trabalho (e não de outra parte do sistema), este aumento da entropia não é
compensado por um decréscimo da entropia em outra parte do sistema.
Exercício sugerido
Considere dois corpos de grandes dimensões (para que a perda ou o ganho de calor não afecte a
sua temperatura), um a 273 K e outro a 373 K. Mostre que é impossível (i.e., a 2ª lei é violada) a
simples transferência de uma pequena quantidade de energia térmica, 8 J, do corpo mais frio para
o mais quente.
Resumindo alguns aspectos importantes do princípio do aumento da entropia, podemos dizer que:
- É um princípio direccional, isto é, limita a direcção em que o processo pode seguir. Um
processo tem de seguir na direcção que obedeça ao princípio do aumento da entropia (um
processo que viole este princípio é impossível);
- A função entropia é uma propriedade não conservativa, isto é, há geração de entropia. Só
nos processos reversíveis (ideais) é que a entropia se conserva, ou seja, os processos
reversíveis não geram entropia. Nos processos reais a entropia aumenta sempre;
- A 2ª lei estabelece que todos os sistemas são capazes de, espontaneamente, atingir um
estado de equilíbrio e que a entropia aumenta continuamente à medida que o estado de
equilíbrio se aproxima. Este é atingido quando a função entropia atinge o valor máximo
compatível com os constrangimentos impostos ao sistema;
- O princípio do aumento da entropia está ligado directamente ao conceito de
irreversibilidade. É a presença de irreversibilidades que leva a entropia a aumentar.
Quanto maiores elas forem, maior será esse aumento: a função entropia pode então servir
para medir os efeitos dissipativos ou perdas associadas a qualquer processo real.
6.5. O que é a entropia?
Vemos que a entropia é uma propriedade útil do ponto de vista da previsão da evolução dos
processos termodinâmicos. Mas o que é a entropia?
O facto de não conseguirmos descrever completamente essa propriedade não lhe retira a
utilidade. A entropia, que começou por ser definida na Termodinâmica, passou a ter uma
importância crescente na Mecânica Estatística em que o comportamento macroscópico de uma
substância é descrito em termos do comportamento estatístico dos átomos e moléculas da
substância. Neste contexto, um dos resultados é que os sistemas isolados tendem para a desordem
e a entropia é uma medida desta desordem. A entropia de uma substância é mínima no estado
sólido em que as moléculas oscilam continuamente em torno das suas posições de equilíbrio mas
não se deslocam umas em relação às outras, sendo as suas posições em qualquer instante
previsíveis com um certo grau de certeza. E é máxima no estado gasoso, fortemente desordenado,
em que as moléculas se movem aleatoriamente, colidem umas com as outras e mudam de
direcção, tornando extremamente difícil prever o estado microscópico em qualquer instante.
Todos os processos físicos tendem para estados mais prováveis do sistema e das suas vizinhanças
e o estado mais provável é sempre um estado de maior desordem. Como a entropia é uma medida
47
TERMODINÂMICA APLICADA
2009 Isabel Ambar
dessa desordem, então isto equivale a dizer que a entropia do Universo aumenta sempre em todos
os processos reais. Este é um modo alternativo de enunciar a 2ª lei da Termodinâmica.
O facto de a entropia de um sistema isolado nunca poder decrescer em qualquer transformação
tem uma interpretação clara do ponto de vista estatístico. Boltzmann provou que a entropia de um
dado estado de um sistema termodinâmico está relacionada com a probabilidade de ocorrência
desse estado. Vamos ver como estabelecer esta relação.
Na teoria cinética dos gases, as moléculas são consideradas partículas a moverem-se de um modo
caótico. Suponhamos que o gás ocupa inicialmente um dado volume Vi e que, por remoção de
uma partição ou parede que o separava de um espaço vazio, ele passa a ocupar um espaço maior,
Vf. A distribuição das moléculas nesse novo espaço pode ser obtida em termos de probabilidade.
Consideremos o instante inicial após se ter retirado a partição, em que as moléculas ainda
ocupam o volume inicial, e vamos estimar a probabilidade dessas moléculas virem a ocupar uma
dada configuração através de movimentos aleatórios num volume maior. Se cada molécula
ocupar um volume microscópico Vm, então o número total de locais possíveis dessa molécula
num volume inicial Vi, é: Wi = Vi/Vm (o que é um número muito grande). Se tivermos N
moléculas, cada uma podendo ir para qualquer dos Vi/Vm locais, o número de possibilidades
V
passará a ser W = ( i ) N . Do mesmo modo, se o volume aumentou para Vf, o número de
i
Vm
estados equivalentes aumenta para W = ( Vf ) N . As probabilidades respectivas são proporcionais
f
Vm
a estes números de possibilidades ( Pi = cWi
probabilidades é:
W
f = ⎛⎜ Vf
⎜V
⎝ i
W
i
Pf = cW ). A razão entre estas
f
e
⎞
⎟
⎟
⎠
N
Aplicando logaritmos naturais a esta equação e multiplicando pela constante de Boltzmann (kB):
kB
⎛V ⎞
W
ln( f ) = k B ln⎜ f ⎟
⎜V ⎟
Wi
⎝ i⎠
N
⎛V
= k B N ln⎜ f
⎜V
⎝ i
⎞
⎛V
⎟ = k n N ln⎜ f
B
A ⎜V
⎟
⎠
⎝ i
⎞
⎟
⎟
⎠
em que n é o número de moles e NA é o número de Avogadro. Como kB=Ru/NA, vem:
V
k B lnWf − k B lnWi = n R u ln( f )
Vi
Por outro lado, se calcularmos a variação da entropia de n moles de um gás ideal numa expansão
livre adiabática de Vi para Vf, obtemos a seguinte expressão (ver Nota):
V
S f − S i = n R u ln( f )
Vi
Comparando as duas expressões, podemos obter a seguinte relação entre entropia e
probabilidade:
S ≡ kB ln W
48
TERMODINÂMICA APLICADA
2009 Isabel Ambar
W é o número de estados microscópicos disponíveis para um sistema num dado estado
macroscópico. Devido à tendência estatística dos sistemas para evoluírem para estados de maior
probabilidade e maior desordem, todos os processos naturais são irreversíveis aumentando a
entropia. A conclusão importante é que a entropia é uma medida da desordem a nível
microscópico. Apesar daquela expressão ter sido deduzida a partir de um exemplo específico (a
expansão livre de um gás ideal), teríamos chegado ao mesmo resultado se tivéssemos seguido um
caminho mais rigoroso e geral.
Como se disse anteriormente, as moléculas de uma substância na fase sólida oscilam
continuamente mas estas oscilações vão enfraquecendo à medida que a temperatura diminui. Em
princípio, as moléculas estariam imóveis no zero absoluto. Este representa então um estado limite
de ordenação molecular (e de energia mínima). Portanto, a entropia de uma substância cristalina
no zero absoluto é nula. Esta é a Terceira Lei da Termodinâmica, a qual fornece um ponto de
referência absoluto para a determinação da entropia. A entropia relativa a este ponto é a entropia
absoluta.
Nota: Uma expansão livre de um gás não é um processo reversível. Para calcular a respectiva
variação de entropia, temos de imaginar um processo reversível equivalente que leve o sistema do
mesmo estado inicial ao mesmo estado final. No caso da expansão livre adiabática de um gás
ideal, como a energia interna não varia, a temperatura também não varia e portanto podemos
imaginar um processo reversível isotérmico que tenha os mesmos estados inicial e final da
expansão livre. Mas para este processo, como U não varia (a energia interna de um gás ideal é
função exclusiva da temperatura), então W + Q = 0. E portanto
ΔS =
∫
(
)
δQ rev 1
V
V
1
1
= Q rev = - Wproc. isot. = nR u T ln( f ) = n R u ln( f )
T
T
T
T
Vi
Vi
7. Ciclos de gás e de vapor
7.1. Ciclos de gás
A maior parte dos dispositivos de produção de potência trabalham em ciclos em que há efeitos
devidos ao atrito e à ausência do tempo suficiente para se estabelecerem condições de equilíbrio
ao longo do ciclo. Retirando todas as irreversibilidades internas (i.e., as irreversibilidades que
ocorrem dentro das fronteiras do sistema durante o processo) e complexidades, obtém-se um
ciclo semelhante ao ciclo real mas composto por processos internamente reversíveis, i.e., um
ciclo ideal.
Estes ciclos, ao contrário do ciclo de Carnot (totalmente reversível), não precisam de ser
externamente reversíveis (i.e., não há ocorrência de irreversibilidades no exterior das fronteiras
do sistema durante o processo), são apenas internamente reversíveis. A eficiência térmica de um
ciclo ideal, em geral, é menor do que a de um ciclo totalmente reversível operando entre as
mesmas temperaturas, mas é consideravelmente superior ao de um ciclo real.
Nos ciclos de gás o fluido permanece no estado gasoso ao longo de todo o ciclo enquanto que nos
ciclos de vapor o fluido existe na fase de vapor em parte do ciclo e na fase líquida em outra parte
do ciclo.
49
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Os ciclos termodinâmicos podem ser classificados em fechados (o fluido volta ao estado inicial
no fim do ciclo e é recirculado) e abertos (o fluido é renovado no fim de cada ciclo; o dispositivo
opera num ciclo mecânico mas o fluido não sofre um processo cíclico).
Os dispositivos de produção de potência podem ser motores de combustão interna (o calor a
fornecer ao fluido é obtido através da combustão do combustível no interior das fronteiras do
sistema) e motores de combustão externa (o calor a fornecer ao fluido vem de uma fonte externa
tal como uma fornalha, um reactor nuclear, o Sol, etc.)
Nos motores de combustão interna, a composição do fluido passa de uma mistura de ar e
combustível para uma mistura de produtos da combustão ao longo de um ciclo. Apesar de os
motores de combustão interna operarem em ciclos mecânicos (o êmbolo volta à sua posição
inicial ao fim de cada ciclo), o fluido não sofre um ciclo termodinâmico completo. Os ciclos de
gás na realidade são muito complexos e na sua análise utilizam-se algumas aproximações –
hipóteses do ar padrão.
Um ciclo de ar padrão é pois um ciclo ideal baseado nas seguintes aproximações: (i) o fluido é
apenas ar, o qual circula em circuito fechado e se comporta como um gás ideal; (ii) todos os
processos são internamente reversíveis; (iii) o processo de combustão é substituído por um
processo de adição de calor a partir de uma fonte externa; (iv) o processo de escape dos produtos
da combustão é substituído por um processo de rejeição de calor para a vizinhança, o qual leva o
fluido ao estado inicial.
Ao aplicar o ciclo de ar padrão, é por vezes costume introduzir constrangimentos adicionais nas
propriedades do ar, como, por exemplo, considerar que os calores específicos (cp e cv) dependem
da temperatura.
Ciclo de Carnot
Comecemos por recordar o ciclo de Carnot. Como vimos, o ciclo é reversível e constituído por 4
processos, sendo dois isotérmicos (T=cte.) e dois adiabáticos (e como são reversíveis, são
processos isentrópicos). A sua representação num diagrama p-V (Clapeyron) e num diagrama T-S
(temperatura-entropia) será então:
p
A
Qq
Qq
T
B
B
W
W
D
D
Qf
A
C
Qf
C
S
V
No diagrama T-S, a área subtendida pela curva representativa de um processo reversível
corresponde à transferência de calor nesse processo:
50
TERMODINÂMICA APLICADA
2009 Isabel Ambar
δQ rev
⇒ δQ rev = T dS ⇒ Qrev = ∫ T dS
T
Como o trabalho realizado durante este ciclo é igual à diferença entre as quantidades de calor
trocadas (Qq e Qf), então ele é proporcional à área envolvida pelo rectângulo ABCDA.
dS =
Consideremos as variações de entropia que ocorrem ao longo de um ciclo de Carnot entre as
temperaturas Tq e Tf. Ao completar um ciclo, o sistema absorveu (mantendo a temperatura Tq)
uma quantidade de calor Qq e cedeu (mantendo a temperatura Tf) uma quantidade de calor Qf . Os
outros dois processos que fazem parte do ciclo, são adiabáticos e portanto não há variação de
entropia do sistema). Então a variação total de entropia é dada por:
Qq Qf
ΔS (cicloCarnot) =
−
Tq
Tf
Mas já vimos que
Qf
T
= f num ciclo de Carnot e portanto: ΔS(cicloCarnot) = 0
Q q Tq
Ciclo de Otto
Vejamos agora um exemplo concreto de uma máquina térmica que é o motor de combustão de
um automóvel. Um motor a gasolina trabalha em ciclos, sendo cada um destes aproximado (não
se consideram os efeitos do atrito, perdas de calor por condução, etc.) pelo ciclo de Otto ideal
(internamente reversível). Este ciclo, ilustrado no diagrama junto, tem 5 fases:
(i) O→A: entrada da mistura de vapor de gasolina e ar no cilindro, à pressão atmosférica:
aumento do volume de V1 para V2;
(ii) A→B: compressão adiabática da mistura de ar e gasolina desde V2 a V1 (trabalho realizado
sobre o sistema) com aumento da temperatura;
(iii) B→C: combustão da mistura quente (devida à faísca de ignição) com adição de uma
quantidade de energia térmica (Qq); os produtos resultantes atingem uma temperatura e uma
pressão muito elevadas mas o volume mantém-se constante (não há trabalho realizado pelo
gás);
(iv) C→D: expansão adiabática dos produtos da combustão, com descida da temperatura e
trabalho realizado pelo gás;
(v) D→A: abre-se uma válvula de escape e liberta-se gás mantendo-se o volume constante;
libertação de energia térmica (Qf ) pelo gás à medida que a sua pressão diminui;
(vi) A→O: os gases residuais são expelidos à pressão atmosférica e V decresce de V2 para V1.
C
p
C
T
Q=0
Qq
V cte
D
B
Qf
B
Qq
O
V1
A
A
V2
D
W
Q=0
V
Qf
V cte
S
51
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Ciclo ideal de Otto:
ar
ar
ar
Compressão
isentrópica
ar
Adição de
calor (V cte)
Expansão
isentrópica
Rejeição de
calor (V cte)
Motor de explosão a 4 tempos:
escape
Compressão
Expansão
mistura
Admissão
Escape
O rendimento de um ciclo de Otto em que se possa considerar a mistura de ar e combustível
como um gás ideal, pode ser calculado aplicando o 1º princípio da Termodinâmica à unidade de
massa:
Δu = q + w
Os dois processos em que há transferência de calor (B-C, D-A) fazem-se a volume constante
(sendo W=0) e portanto a respectiva transferência de calor é igual à variação de energia interna
do gás ideal (para o qual u = cv T, sendo cv o calor específico a volume constante):
Qq= uC – uB = cV (TC – TB)
Qf= uA – uD = cV (TA – TD)
Por outro lado, vamos usar a seguinte relação, válida num processo adiabático reversível
(isentrópico) de um gás ideal:
T Vγ-1 = constante
em que γ (>1) é a razão entre os calores específicos a pressão constante e a volume constante
(coeficiente adiabático: γ = cp/cv)
TA ⎛ VB ⎞
⎟
=⎜
TB ⎜⎝ VA ⎟⎠
γ −1
⎛V ⎞
= ⎜⎜ C ⎟⎟
⎝ VD ⎠
γ −1
T
= D
TC
T
⇒ D =
TA
TC
TB
O rendimento do ciclo de Otto é dado por:
52
TERMODINÂMICA APLICADA
2009 Isabel Ambar
η Otto =
Wres
Qq
=
Qq − Qf
= 1−
Qq
⎛V
T
η Otto = 1 − A = 1 − ⎜⎜ B
TB
⎝ VA
⎞
⎟⎟
⎠
Qf
c (T - T )
(T - T )
= 1− V D A = 1− D A
c V (TC - TB )
(TC - TB )
Qq
γ −1
= 1−
η Otto = 1 −
T
TA ( D − 1)
TA
= 1−
T
TB ( C − 1)
TB
1
⎛ VA
⎜⎜
⎝ VB
1
⎛ V2
⎜⎜
⎝ V1
⎞
⎟⎟
⎠
γ −1
⎞
⎟⎟
⎠
γ −1
== 1 −
1
r γ −1
= 1−
TA
T
= 1− D
TB
TC
em que VA/VB = Vmáx/Vmín = V2/V1 é a razão de compressão (r). Quanto maior for esta razão,
maior é o rendimento. Com uma razão de compressão de valor típico 8 e γ = 1,4, o rendimento
teórico é de 56%, o que é muito maior que o rendimento em motores reais (25% a 30%), porque
nestes há sempre os efeitos dissipativos associados ao atrito, às perdas de calor através das
paredes do cilindro e à combustão incompleta da mistura ar+gasolina. Os motores Diesel têm
rendimentos maiores porque têm uma compressão maior e, portanto, a temperatura de combustão
é maior.
Rendimento do ciclo ideal de Otto em função da razão de compressão r, para γ=1,4
(Çengel & Boles, 2006)
Da figura vemos que o aumento do rendimento com a razão de compressão se torna menos
pronunciado para valores elevados de r. Quando são utilizadas razões de compressão elevadas a
temperatura da mistura ar+gasolina pode ultrapassar a temperatura de auto-ignição da gasolina
(esta entra em ignição mesmo sem a faísca da vela) durante o processo de combustão, levando à
queima antecipada e rápida da gasolina e produzindo um ruído audível. Para evitar este efeito
prejudicial do desempenho do motor convém não exceder certos limites da razão de compressão.
Para melhorar o desempenho de um motor a gasolina utilizando valores elevados (até cerca de
12) da razão de compressão sem o perigo da auto-ignição, recorreu-se à mistura de outras
53
TERMODINÂMICA APLICADA
2009 Isabel Ambar
substâncias (tal como o chumbo) com a gasolina para aumentar o índice de octana, uma medida
da resistência à auto-ignição. No entanto, esta adição foi proibida a partir de meados dos anos
1970s, no combate à poluição do ambiente e aos problemas de saúde causados pelo chumbo.
Hoje em dia, o acesso a combustíveis de elevado índice de octana e a melhoria do desempenho
dos automóveis (menos pesados, mais aerodinâmicos) melhorou consideravelmente a eficiência
dos motores.
O outro parâmetro de que depende o rendimento é o coeficiente adiabático (γ). Para um dado r, o
ciclo ideal de Otto utilizando um gás monoatómico (por exemplo, árgon ou hélio, γ = 1,667) tem
o máximo do rendimento. O gradiente adiabático decresce à medida que as moléculas do fluido
são maiores (para o ar γ = 1,4, para o dióxido de carbono γ = 1,3) e portanto o rendimento
também decresce. O rendimento dos motores reais de ignição por faísca está entre 25% e 30%.
Variação do rendimento do ciclo de Otto com o coeficiente adiabático (γ) do fluido
(adaptado de Çengel & Boles, 2006)
Ciclo de Diesel
Há diferenças importantes entre um motor a gasolina e um motor a gasóleo (motor Diesel). Neste
último, em vez da entrada inicial de ar e combustível, só entra ar. Este vai aquecer por
compressão adiabática até que a sua temperatura seja suficientemente alta para provocar a ignição
do óleo que entretanto, após a compressão, é injectado (como “spray”) no cilindro. A taxa de
fornecimento de óleo é regulada de modo que a combustão se dê quase isobaricamente (o êmbolo
vai sendo empurrado durante a combustão). O resto do ciclo é igual ao do motor a gasolina (ciclo
de Otto). O ciclo de Diesel é o ciclo ideal (internamente reversível) para motores de ignição por
compressão e está representado na figura seguinte, tanto num diagrama p-V como num diagrama
T-S.
54
TERMODINÂMICA APLICADA
2009 Isabel Ambar
p
B
Qq
Qq
C
T
Q=0
B
C
p cte
D
Q=0
D
Qf
V cte
A
A
O
V2
V1
V3
Qf
V
S
O rendimento de um ciclo ideal de Diesel é dado pela expressão:
η Diesel =
Wres
Qq
=
Qq − Qf
Qq
T
TA ( D − 1)
Qf
(TD - TA )
TA
1
= 1−
= 1−
= 1−
= 1−
γ
T
γ (TC - TB )
Qq
r −1
γ TB ( C − 1)
TB
⎡ r γ −1 ⎤
⎥
⎢ c
⎢⎣ γ(rc − 1) ⎥⎦
VC V3
=
, é a razão entre o volume dos cilindros depois (V3) e antes (V2) do
VB V2
processo de combustão (razão de admissão) e r é a razão de compressão definida anteriormente
V
V
( r = máx = 1 ).
Vmín V2
em que rc =
Nota
Utilizaram-se as seguintes igualdades para deduzir a expressão final do rendimento η Diesel :
-
TV γ −1 = constante para uma transformação adiabática de um gás ideal:
TA VA γ −1 = TB VB γ −1 , então:
-
TA ⎛ VB ⎞
⎟
=⎜
TB ⎜⎝ VA ⎟⎠
γ −1
=
1
r γ −1
e portanto:
TA
1
=
TB r γ −1
pV γ = constante para uma transformação adiabática de um gás ideal:
p A VA γ = p B VB γ e p C VC γ = p D VD γ . Dividindo membro a membro estas 2 igualdades e
sabendo que pB = pC e VA = VD, vem:
-
⎛ VC
⎝ VB
(rC )γ = ⎜⎜
γ
⎞
p
⎟⎟ = D (i)
pA
⎠
p T-1 = constante para uma transformação isocórica de um gás ideal:
p D TD
=
(ii)
p A TA
Combinando (i) com (ii), vem:
•
TD
= (rC )γ
TA
T V-1 = constante para uma transformação isobárica de um gás ideal:
T
TC VC
=
= rC Portanto C = rC
TB
TB VB
55
TERMODINÂMICA APLICADA
2009 Isabel Ambar
⎡ r γ −1 ⎤
Os rendimentos do ciclo de Otto e do ciclo de Diesel diferem pelo factor ⎢ c
⎥ . Como esta
⎢⎣ γ(rc − 1) ⎥⎦
quantidade é sempre maior que 1, então: ηOtto > ηDiesel quando os dois ciclos operam com a
mesma razão de compressão. No entanto, como os motores a Diesel podem trabalhar com
compressões muito maiores, eles acabam por ser mais eficientes (na realidade, podem atingir
valores entre 30% e 40%). No caso limite de rc = 1, aquele factor é igual a 1 e os rendimentos dos
2 ciclos seriam iguais.
Variação do rendimento do ciclo de Diesel em função da razão de compressão (r) e da razão de admissão
(rc) (adaptado de Çengel & Boles, 2006)
Actividade sugerida
Para ter uma ideia do modo como trabalham os motores a gasolina e a Diesel e as suas diferenças,
consulte os seguintes sítios da net: http://www.howstuffworks.com/diesel1.htm;
http://www.howstuffworks.com/engine1.htm
Ciclos de Stirling e de Ericsson
Tanto o ciclo ideal de Otto como o de Diesel são compostos por processos internamente
reversíveis e portanto são ciclos internamente reversíveis. Mas não são ciclos totalmente
reversíveis porque incluem transferências de calor entre corpos com diferenças finitas de
temperatura, isto é, há adição ou rejeição de calor em processos não-isotérmicos (para que dois
corpos interajam reversivelmente, isso implica estarem em equilíbrio térmico um com o outro,
i.e., terem igual temperatura). Isso quer dizer que o rendimento desses ciclos terá de ser sempre
inferior ao do ciclo de Carnot que opere entre as mesmas temperaturas.
Há dois outros ciclos que envolvem processos isotérmicos de adição e de rejeição de calor: o
ciclo de Stirling e o de Ericsson. Diferem do ciclo de Carnot em que os dois processos
isentrópicos deste são substituídos por dois processos a volume constante (Stirling) ou a pressão
constante (Ericsson). Ambos os ciclos utilizam um processo de regeneração em que há, durante
parte do ciclo, transferência de calor para um dispositivo de depósito de energia térmica
(regenerador), o qual é devolvido ao fluido durante a outra parte do ciclo.
Robert Stirling (1790-1878) propôs um ciclo composto por 2 processos isotérmicos reversíveis e
2 processos isocóricos (a volume constante) reversíveis:
56
TERMODINÂMICA APLICADA
2009 Isabel Ambar
(i) A→B: Expansão isotérmica do gás em contacto térmico com um reservatório à temperatura
Tq; o gás absorve a quantidade de calor Qq de um reservatório à temperatura Tq e realiza um
trabalho WAB ao empurrar o êmbolo;
(ii) B→C: é extraído calor do fluido para o regenerador, a volume constante, até a temperatura do
fluido atingir Tf.
(iii) C→D: Compressão isotérmica até ao volume inicial, sendo cedido calor a um reservatório à
temperatura Tf.
(iv) D→A: O fluido recebe calor do regenerador, a volume constante, até atingir o estado inicial.
p
Qq
D
Qq
T
A
Tq
REG
V cte
B
Tf
Tf
Qf
B
A
Tq
REG
D
Qf
C
V cte
C
S
V
O ciclo trabalha entre dois reservatórios a temperaturas constantes se as quantidades de calor
trocadas nos processos B-C e D-A puderem ser conservadas dentro do sistema. Elas são iguais
em módulo e o que será preciso é arranjar maneira de guardar a quantidade de energia térmica
cedida durante o processo B-C e depois fornecer a mesma quantidade de energia térmica ao
fluido durante o processo D-A. Para tal é necessário utilizar um regenerador, que pode ser
considerado como uma série de reservatórios de calor cujas temperaturas variam
diferencialmente. Assim as transferências de calor com esses reservatórios são sempre
reversíveis.
Para compreender a realização de um ciclo de Stirling num sistema fechado considere-se um
cilindro com dois êmbolos e um regenerador no meio destes. O regenerador pode ser uma malha
de cerâmica ou de metal ou qualquer espécie de tampão poroso com grande inércia térmica que é
utilizado para o armazenamento temporário de energia térmica.
Inicialmente, a câmara da esquerda contém todo o gás, o qual está a alta temperatura (Tq) e
pressão.
(i) Durante o processo A-B, é transferido calor para o gás a partir de um reservatório de calor à
temperatura Tq. À medida que o gás se expande isotermicamente, o êmbolo da esquerda movese para fora, realizando trabalho, e a pressão do gás diminui.
(ii) Durante o processo B-C, ambos os êmbolos movem-se para a direita em paralelo (para manter
o volume constante) até que todo o gás é forçado a passar para a câmara da direita. À medida
que o gás atravessa o regenerador, transfere calor para este e a temperatura desce para Tf. Para
que esta transferência de calor seja reversível, a diferença de temperatura entre o gás e o
regenerador deve ser infinitesimal em qualquer altura. Assim, a temperatura do regenerador
deve ser Tq do lado esquerdo e Tf do lado direito quando se atinge o estado C.
57
TERMODINÂMICA APLICADA
2009 Isabel Ambar
(iii) Durante o processo C-D, o êmbolo da direita move-se para dentro comprimindo o gás. Há
transferência de calor do gás para um reservatório de calor à temperatura Tf, de modo que a
temperatura do gás se mantém constante (e igual a Tf) enquanto a pressão aumenta.
(iv) No processo D-A, ambos os êmbolos se deslocam para a esquerda à mesma velocidade (gás a
volume constante), forçando o gás a ocupar a câmara do lado esquerdo. A temperatura do gás
aumenta de Tf para Tq quando atravessa o regenerador, reavendo a energia térmica armazenada
durante o processo B-C.
Qf
Qq
A⇒B
B⇒C
C⇒D
D⇒A
O processo A-B (ou C-D) corresponde à transferência de calor por um processo isotérmico
reversível (Q=TΔS) e portanto podemos escrever: Qq = Tq (SB – SA)>0 (ou Qf = Tf (SD – SC)< O).
O rendimento térmico teórico de um ciclo de Stirling é então dado por:
Qq − Qf
Tq (S B − S A ) − Tf (SC − S D )
=
ηStirling =
Tq (S B − S A )
Qq
Como a transferência de calor para o regenerador é reversível, a variação de entropia do fluido
tem de ser igual à do regenerador, em qualquer dos processos B-C ou D-A. Mas a variação de
entropia do regenerador durante um ciclo é nula visto que volta ao estado inicial.
Consequentemente, (SC – SB) + (SA-SD) = 0 para o fluido. Por outro lado, sabemos que a variação
de entropia do fluido no ciclo completo é nula e portanto:
(SA – SB) + (SB-SC) + (SC – SD) + (SD-SA) = 0
Atendendo à condição anterior (SC – SB) + (SA-SD) = 0, terá então de ser: (SA – SB) + (SC – SD) =
0
(SB-SA) = (SC-SD)
Substituindo na expressão do rendimento:
ηStirling =
Tq − Tf
T
= 1− f
Tq
Tq
O rendimento teórico do ciclo de Stirling é igual ao do ciclo de Carnot, o que não é de admirar
visto que considerámos o ciclo de Stirling totalmente reversível e portanto o seu rendimento é
igual ao de qualquer ciclo reversível (tal como o de Carnot) trabalhando entre as mesmas
temperaturas.
O ciclo de Ericsson é semelhante ao de Stirling exceptuando os dois processos a volume
constante (isocóricos) que são substituídos por processos a pressão constante (isobáricos).
58
TERMODINÂMICA APLICADA
2009 Isabel Ambar
p
D
Tq
Qq
B
A
Tq
Tf
Qf
Qq
T
A
p cte
Tf
C
p cte
D
C
Qf
B
S
V
A figura seguinte mostra um sistema com um escoamento estável a operar num ciclo de Ericsson.
A compressão e a expansão isotérmicas são realizadas, respectivamente, num compressor e numa
turbina. Os escoamentos de fluido quente e de fluido frio entram no regenerador em extremos
opostos dando-se a troca de calor entre esses dois escoamentos.
REGEN.
compr.
turbina
W
Qf
Qq
Considerando este ciclo totalmente reversível (o que implica que as transferências de calor têm
de ser feitas através de uma diferença de temperatura infinitesimal em todas as componentes do
ciclo, incluindo o regenerador), então o seu rendimento deverá ser igual ao do ciclo de Carnot e
ao do ciclo de Stirling operando entre os mesmos limites de temperatura (Tq e Tf):
T
η Ericsson = ηStirling = η Carnot = 1 − f
Tq
Na realidade, não só todos os processos de transferência de calor se fazem através de uma
diferença finita de temperaturas, como o regenerador não é 100% eficiente e há quedas de
pressão ao longo do circuito do fluido.
Tem surgido recentemente interesse em desenvolver motores de Stirling ou de Ericsson para
camionetas, autocarros e mesmo automóveis mas ainda estão longe de competir com os motores a
gasolina ou a diesel. Ambos são motores de combustão externa (o combustível é queimado fora
dos cilindros) ao contrário dos motores a gasolina ou a diesel em que a combustão é interna. Os
motores de combustão externa têm algumas vantagens, tais como haver vários combustíveis que
podem ser usados para fornecer energia térmica, o processo de combustão ser mais completo
(menor poluição do ar e melhor aproveitamento do combustível) e operarem em ciclos fechados
(o que leva a escolher como fluido circulante um gás estável, quimicamente inerte, com alta
59
TERMODINÂMICA APLICADA
2009 Isabel Ambar
condutividade térmica). Apesar das limitações práticas dos motores de Stirling e de Ericsson, eles
mostram que o processo de regeneração pode aumentar a eficiência das máquinas. O ciclo de
Brayton, que vamos ver a seguir, tem algumas semelhanças com o de Ericsson.
Ciclo de Brayton
O ciclo de Brayton foi inicialmente proposto por George Brayton por volta de 1870. Hoje em dia
é utilizado em turbinas a gás apenas quando tanto a compressão como a expansão têm lugar em
máquinas que rodam. As turbinas de gás geralmente operam em ciclo aberto mas podemos
aproximar o seu funcionamento através de um ciclo fechado, o ciclo de Brayton.
No ciclo de Brayton, os processos de compressão e expansão são adiabáticos e os processos de
adição e remoção de calor ocorrem a pressão constante (isobáricos).
Qq
p
p2
B
C
Qq
T
C
p cte
B
Q=0
D
Q=0
p1
A
Qf
A
D
p cte
V
Qf
S
O rendimento do ciclo de Brayton é dado pela seguinte expressão:
η Brayton = 1 −
1
( γ −1 )
γ
rp
p
em que rp = 2 é a razão de pressão.
p1
Resumo dos ciclos de gás:
Ciclo
Proc. isotérmico
Proc. adiabático
Carnot
Otto
Diesel
Stirling
Ericsson
Brayton
2 (transf. calor)
2
2
2
2 (transf. calor)
2 (transf. calor)
Proc. isocórico
Proc. isobárico
2 (transf. calor)
1 (transf. calor)
2
1 (transf. calor)
2
2 (transf. calor)
2
60
TERMODINÂMICA APLICADA
2009 Isabel Ambar
7.2. Ciclos de vapor
No capítulo 11 do curso, ao falar das propriedades físicas de uma substância pura, voltaremos ao
tema dos ciclos para exemplificar os processos que têm lugar ao longo de um ciclo de vapor.
8. Energia disponível. Trabalho máximo. Irreversibilidade.
A primeira lei permite relacionar as várias formas de energia em termos das suas quantidades mas
a mesma quantidade de energia sob diferentes formas pode ter utilidades diferentes para a
sociedade. Quer dizer, na energia a qualidade é importante, para além da quantidade. A 2ª lei
fornece os métodos para aferir a qualidade da energia.
8.1 Rendimento máximo de uma máquina térmica
Sempre que se dá um processo irreversível, a entropia do universo aumenta – há geração de
entropia. Na operação real de um dispositivo tal como um motor ou um frigorífico, em geral é
possível calcular a soma de todas as variações de entropia. O facto desta soma ser positiva
permite-nos tirar conclusões úteis sobre o desempenho desse dispositivo. Vejamos o exemplo de
uma máquina térmica que perfaz um ciclo arbitrário, ao longo do qual extrai calor Qq de um
reservatório a Tq, produzindo trabalho W e rejeitando calor ⎜Qf ⎜= ⎜Qq⎜ - ⎜W⎜ para um
reservatório à temperatura Tf. A variação de entropia do universo é:
ΔS univ = ∑ ΔS =
Qq − W
Tf
−
Qq
Tq
≥0
Então o trabalho realizado será W ≤ Q q (1 − Tf )
Tq
O trabalho máximo é:
Wmáx = Q q (1 −
Como
Wmáx
Qq
Tf
)
Tq
é o rendimento máximo de uma máquina térmica trabalhando entre os dois
reservatórios, então
η máx = 1 −
Tf
= rendimento de uma máquina de Carnot.
Tq
O rendimento máximo de qualquer máquina operando entre dois reservatórios, é o de uma
máquina de Carnot.
Suponhamos que uma quantidade de calor Q pode ser extraída de um reservatório à temperatura
T e que se pretende converter o máximo possível desta energia térmica em trabalho. Se To for a
temperatura do reservatório mais frio de que se possa dispor (quanto menor for a temperatura da
fonte fria, maior será o rendimento), então:
T
Wmáx = Q (1 − o )
T
61
TERMODINÂMICA APLICADA
2009 Isabel Ambar
o que representa a quantidade máxima de energia disponível para converter em trabalho, quando
se extrai uma quantidade de energia térmica Q de um reservatório à temperatura T.
8.2. Função disponibilidade
A tendência para os sistemas aumentarem a sua entropia pode ser aproveitada para a realização
de trabalho útil. Consideremos um sistema A que está em contacto térmico e mecânico com um
reservatório R (a parede que os liga é diatérmica e móvel), caracterizado por ter temperatura
constante (To) e pressão constante (po). Um bom exemplo de um reservatório deste tipo é a
própria atmosfera.
Vamos supôr que só há interacções do tipo calor com a atmosfera. O sistema A+R forma um
sistema isolado.
Considerando um processo em que o sistema A recebe uma quantidade de calor Q do
reservatório, a variação de entropia deste é:
A
Q
Q
ΔS R = −
To
R
To, po
A variação de entropia do universo será:
Q
ΔS univ = ΔS A −
≥0
To
Se nesse processo o volume de A variar ∆VA, então a energia interna de A varia ∆UA = Q po∆VA, ou seja ⎜Q⎜= ∆UA+ po∆VA. Substituindo na expressão de ∆Suniv:
ΔSuniv = ΔSA −
ou seja:
ΔUA + poΔVA
To
≥0
To ΔS A − ΔU A − p o ΔVA
≥0
To
e como To>0:
To ΔS A − ΔU A − p o ΔVA ≥ 0
Como po e To são constantes:
-∆(UA-ToSA+poVA)≥0
Vamos definir função disponibilidade como:
D = U – To S + po V
Note-se que a função disponibilidade não é uma função de estado do sistema. É uma propriedade
do conjunto sistema+reservatório.
Então vemos que a variação da função disponibilidade é menor ou igual a zero:
ΔD≤0
62
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Esta desigualdade é equivalente a dizermos que ΔSuniv≥0. Em processos reversíveis, ΔD=0. Em
processos naturais (não reversíveis), a função disponibilidade só pode diminuir (equivalente a
dizer que a entropia de um sistema isolado só pode aumentar).
ΔS univ =
− ΔD
≥0
To
ΔD = -To ΔSuniv ≤ 0
em que ΔSuniv corresponde à entropia gerada.
Já sabemos que no estado de equilíbrio mais estável, a entropia toma o valor máximo compatível
com os constrangimentos. Então para esse estado, a soma da entropia do conjunto
sistema+reservatório será máxima e, portanto, a função disponibilidade será mínima (Dmin).
Para um dado estado do conjunto sistema+reservatório, a grandeza energia utilizável ou energia
disponível do sistema é dada por:
Edisp = D - Dmin ≥ 0
ΔEdisp = ΔD
No estado de equilíbrio mais estável do conjunto A+R:
ΔEdisp = 0.
Se para um dado estado, ΔEdisp>0, o estado diz-se vivo. Se ΔEdisp=0, o estado diz-se morto
(corresponde ao estado de equilíbrio mais estável).
8.3. Princípio do trabalho máximo
O conceito de energia disponível é muito útil em aplicações práticas de engenharia. É um modo
de medir o potencial de um processo para produzir trabalho. Ou de medir a perda de capacidade
de produção de trabalho. Para estabelecer um critério que permita saber o valor máximo do
trabalho que um sistema pode fornecer, vamos calcular o trabalho útil máximo que um sistema,
ao passar de um estado de equilíbrio para outro, pode realizar enquanto troca calor apenas com o
ambiente local. Vamos supôr novamente o conjunto do sistema+reservatório, o que corresponde a
supôr um sistema rodeado por uma atmosfera local com temperatura e pressão constantes. Como
o sistema e a atmosfera não estão em equilíbrio, estas duas regiões podem interagir e produzir
trabalho.
Suponhamos que o conjunto sistema+atmosfera está agora em contacto com um reservatório de
trabalho (RW). Vamos determinar o trabalho máximo útil que pode ser fornecido pelo conjunto
sistema+atmosfera quando o sistema passa de um estado de equilíbrio para outro. Por hipótese, e
numa primeira aproximação, vamos supôr que as interacções do tipo calor só se dão com a
atmosfera (reservatório R).
Consideremos uma variação do estado de A+R, em que o único efeito exterior é a produção de
trabalho (W), o qual é fornecido ao reservatório de trabalho (RW). Por definição, um reservatório
de trabalho é um sistema envolvido por uma parede adiabática impermeável e em que os
processos são quase-estáticos.
63
TERMODINÂMICA APLICADA
2009 Isabel Ambar
A
Q
R
W
RW
To, po
Então a variação de entropia do universo é dada por:
ΔSA + ΔSR+ ΔSRW ≥ 0
Então ΔS A −
em que
Q
To
e ΔSRW = 0
Q
≥0
To
Mas como ΔUA = ⏐Q⏐ + Wtotal, temos:
E portanto:
ΔS A −
ΔS R = −
⏐Q⏐ = ΔUA - Wtotal
ΔU A − Wtotal
≥0
To
ou seja:
Wtotal ≥ Δ (UA – To SA)
Se o sistema estiver a fornecer trabalho à fonte reversível de trabalho e à atmosfera, então o
trabalho total é negativo e portanto: Wtotal= - |Wtotal|
- |Wtotal| ≥ Δ (UA – To SA)
|Wtotal| ≤ - Δ (UA – To SA)
A igualdade só se verifica se o processo for reversível. Nesse caso:
|Wtotal,máx| = - Δ (UA – To SA)
Mas o trabalho total pode ser decomposto em duas fracções: uma correspondente ao trabalho
realizado na variação de volume ΔVA contra a atmosfera (à pressão po) e outra, o trabalho útil
(Wu), que corresponde ao trabalho transferido para o reservatório de trabalho:
Wtotal = -po ΔVA + Wu
Então:
|Wtotal| = po ΔVA + |Wu | ≤ - Δ (UA – To SA)
|Wu| ≤ - Δ (UA – To SA+poVA)
|Wu|≤ - ΔD
Se o processo for reversível, a igualdade verifica-se:
|Wu,máx| = - ΔD
64
TERMODINÂMICA APLICADA
2009 Isabel Ambar
O teorema do trabalho máximo pode ser enunciado do seguinte modo:
Para uma dada variação de estado de um sistema (i.e., para determinados valores de ΔU,
ΔV, ΔS) em presença de um reservatório a p e T constantes, o valor absoluto do trabalho
útil máximo que pode ser recolhido num reservatório de trabalho corresponde a um
processo reversível e é igual ao decréscimo da função disponibilidade.
Nos casos em que o reservatório de trabalho fornece trabalho ao sistema (Wu>0):
como Wtotal ≥ Δ (UA – To SA) e Wtotal = -po ΔVA + Wu então -po ΔVA + Wu ≥ Δ (UA – To SA)
Ou seja:
Wu ≥ ΔD
Então, neste caso, será:
Wu,min = Wu,rev = ΔD
Wtotal,min = Wtotal rev = Δ (UA – To SA)
8.4. Irreversibilidade
A grandeza
I = |Wu,máx| - |Wu| ≥ 0
É a medida da irreversibilidade ou, simplesmente, irreversibilidade. Representa a energia
dissipada ou degradada no processo, isto é, a energia que passou de uma forma
macroscopicamente organizada (e portanto totalmente utilizável para fins úteis) para uma forma
macroscopicamente desorganizada (energia interna de um sistema ou reservatório).
I = |Wu,máx| - |Wu| = - ΔD- |Wu| = -Δ (UA – To SA+poVA) - |Wu| = -ΔUA+ To ΔSA - po ΔVA- |Wu|
Mas ΔUA = ⏐Q⏐ - po ΔVA- |Wu| e ⏐Q⏐ = -To ΔSR e portanto - ΔUA = To ΔSR + po ΔVA+ |Wu|
Então:
I = To (ΔSA + ΔSR) = To ΔSuniv ≥ 0
A medida da irreversibilidade está directamente relacionada com a geração de entropia. A
irreversibilidade só é nula quando não há geração de entropia, i.e., quando o processo é
reversível.
O sistema troca calor não só com a atmosfera mas com outro reservatório
Até agora só se considerou o sistema a trocar calor com a atmosfera. Em casos mais gerais, o
sistema pode trocar calor também com outro reservatório a uma temperatura TR. Vejamos a
forma que neste caso tomam as expressões deduzidas anteriormente. O reservatório de
temperatura RT, ao trocar calor com o sistema vai sofrer uma variação de entropia. Vamos supôr
que o sistema recebe calor QR do reservatório RT.
65
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Q
QR
A
W
R To, po
RT
RW
Então a variação de entropia do universo passa a ser:
ΔSA + ΔSR+ ΔSRW + ΔSRT ≥ 0
ΔS A −
Q
−
QR
≥0
To
TR
ΔUA = |Q| + |QR| +Wtotal, |Q| = ΔUA - |QR| - Wtotal
ΔU A − Q R - Wtotal Q R
ΔS A −
−
≥0
To
TR
Wtotal ≥ −To ΔS A + ΔU A − Q R +
QR
TR
To
Como Wtotal= -|Wtotal| então:
T
Wtotal ≤ −ΔU A + To ΔSA + Q R (1 − o )
TR
T
Wtotal , máx = −ΔU A + To ΔSA + Q R (1 − o )
TR
Como Wtotal = -po ΔVA + Wu, então:
QR
- p o ΔVA + Wu ≥ −To ΔS A + ΔU A − Q R +
To
TR
T
Wu ≤ −Δ(U A + To S A + p o VA ) + Q R (1 − o )
TR
9. Relações termodinâmicas formais. Aplicações à termodinâmica da atmosfera e da água do
mar.
O 1º princípio da Termodinâmica permite calcular a variação de energia interna num processo a
partir da seguinte expressão: dU = đQ + đW
Se o processo for quase-estático reversível, e se a única forma de trabalho for a mecânica, então
podemos escrever:
dU = T dS – p dV
66
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Mas uma vez que todas as grandezas que figuram nesta relação são funções de estado, ela é
válida para todos os processos infinitesimais, reversíveis ou irreversíveis, que relacionam estados
de equilíbrio de sistemas simples.
Nota
Só em processos reversíveis TdS e – pdV representam, respectivamente, o calor e o trabalho em
jogo; em processos irreversíveis, TdS > đQ (ver desigualdade de Clausius) e |- pdV| > |đW|
A partir daquela relação, vamos introduzir várias relações formais as quais permitem numerosas
aplicações.
9.1. Relação fundamental e equações de estado
A relação dU = T dS – p dV que é aplicável no caso de um sistema fechado (i.e., com paredes
impermeáveis, que impedem o fluxo de matéria) sugere a existência de uma função U que
depende de S e de V, do tipo:
U = U(S, V)
Uma equação deste tipo designa-se por relação fundamental. Uma relação fundamental tem de
obedecer a um conjunto de condições, como veremos depois. Se soubermos qual a sua expressão
temos toda a informação termodinâmica sobre o sistema Claro que estamos a falar de sistemas
simples com um único componente. Veremos depois a generalização da relação fundamental para
sistemas com vários componentes.
Naquela equação, as variáveis S e V são as variáveis naturais de U.
Diferenciando a função U(S, V) vem:
⎛ ∂U ⎞
⎛ ∂U ⎞
dU = ⎜
⎟ dV
⎟ dS + ⎜
⎝ ∂V ⎠S
⎝ ∂S ⎠V
Comparando esta expressão da diferencial de U com dU = T dS – p dV, conclui-se que:
⎛ ∂U ⎞
⎜
⎟ =T
⎝ ∂S ⎠ V
⎛ ∂U ⎞
⎜
⎟ = −p
⎝ ∂V ⎠ S
Então as variáveis intensivas T e –p dizem-se variáveis conjugadas, respectivamente, das
variáveis S e V, no esquema (ou na representação) da energia. Como já tínhamos definido logo
no início do curso, duas variáveis, uma extensiva (X) e a outra intensiva (P), são conjugadas entre
si numa representação energética se o produto PdX corresponder a uma quantidade infinitesimal
de energia.
A temperatura e a pressão são derivadas parciais de uma função (U) que depende de S e de V, e
portanto, também são funções de S e de V. Então temos as seguintes relações funcionais:
T = T (S, V)
p = p (S, V)
67
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Estas relações que exprimem os parâmetros intensivos em função de parâmetros extensivos
independentes, denominam-se equações de estado. O conhecimento de uma única equação de
estado não nos permite obter toda a informação termodinâmica sobre o sistema. Só o
conhecimento de todas as equações de estado do sistema é equivalente ao conhecimento da sua
relação fundamental e, consequentemente, a toda a informação termodinâmica sobre o sistema.
Podemos agora fazer um raciocínio semelhante numa representação (ou esquema) entrópica. Para
tal, vamos resolver a relação U=U(S, V) em ordem a S:
S = S(U, V)
Esta é uma relação fundamental mas agora na representação entrópica. A sua diferenciação
permite escrever:
⎛ ∂S ⎞
⎛ ∂S ⎞
dS = ⎜
⎟ dU + ⎜
⎟ dV
⎝ ∂U ⎠ V
⎝ ∂V ⎠ U
Resolvendo dU = T dS – p dV em ordem a dS:
dS =
1
p
dU + dV
T
T
Comparando as duas últimas expressões, obtemos:
1
∂S
( )V =
T
∂U
p
∂S
( )U =
∂V
T
1
p
e
são variáveis intensivas conjugadas, respectivamente, das variáveis
As variáveis
T
T
extensivas U e V, no esquema da entropia. Neste caso, o produto de uma variável intensiva pela
diferencial da extensiva conjugada corresponde a uma quantidade infinitesimal de entropia.
Exemplo de uma relação fundamental (no esquema da energia)
Relação fundamental para um gás ideal:
−nR / C
S − So
⎛ V ⎞ u v ( Cv )
U = U o ⎜⎜ ⎟⎟
e
⎝ Vo ⎠
Derivando esta equação em ordem a V (mantendo S constante) obtemos a expressão de -p, e
derivando em ordem a S (mantendo V constante) obtemos a expressão de T. Eliminando a
variável entropia entre cada uma destas expressões e a relação fundamental, obtêm-se as
seguintes expressões:
nR U
U
p = p (U , V ) = u
e
T = T (U , V ) =
C vV
Cv
Eliminando U entre estas duas equações obtém-se a equação de estado dos gases ideais:
p=
nRu T
V
Aquela primeira expressão permite escrever U = U(V, T) = Cv T, relação que também já
conhecemos. De facto, toda a informação termodinâmica sobre o sistema está contida na relação
fundamental.
68
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Se agora considerarmos um sistema aberto, em que o fluxo de matéria através das suas paredes é
permitido, vamos ver qual a forma que as expressões anteriores vão assumir.
Supondo um sistema com C componentes independentes, a expressão da variação da energia
interna entre dois estados de equilíbrio tem agora um termo suplementar que corresponde ao
fluxo de matéria através da fronteira:
C
∂U
dU = TdS − pdV + ∑ (
) dn i
n
∂
i
i =1
em que n1, n2, ..., nC representam os números de moles dos C componentes independentes.
Definindo o potencial electroquímico ou, simplesmente, potencial químico do componente i
como:
μi = (
∂U
)S, V,n j
∂n i
A expressão de dU toma a forma:
C
dU = TdS − pdV + ∑ μ idn i
i =1
A relação fundamental terá a forma:
U = U(S, V, ni) = U(S, V, n1, …, nC)
Nota
É sempre possível exprimir a energia interna em função de parâmetros sem ser exclusivamente S,
V e ni, por exemplo: U = U(T, V, ni). Mas esta relação não seria uma relação fundamental e não
conteria toda a informação termodinâmica sobre o sistema!
As equações de estado são as seguintes:
T = T (S, V, ni)
p = p (S, V, ni)
μi = μi (S, V, ni)
Se quisermos passar para o esquema da entropia, a relação fundamental será:
S = S (U, V, ni)
E agora teremos:
1
∂S
( ) V,n i =
T
∂U
p
∂S
( ) U,n i =
T
∂V
μ
∂S
(
) U,V,n j = − i
T
∂n i
9.2. Relação de Euler e relação de Gibbs-Duhem
69
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Uma função de uma ou mais variáveis f(x1, x2, ...) diz-se homogénea de grau k, se obedecer à
seguinte condição:
f(λx1, λx2, ...) = λk f(x1, x2, ...)
em que λ é um parâmetro arbitrário. O teorema de Euler diz o seguinte: para uma função
homogénea de grau k, a soma dos produtos das variáveis pelas derivadas da função em ordem a
essas variáveis é igual ao produto do grau de homogeneidade pela função. Nesta caso seria:
∑
xi (
∂f
) x = k f ( x1 , x2 ,...)
∂x i j
Dado que as variáveis U, S, V, e ni são todas variáveis extensivas (aditivas em relação à massa),
se a massa do sistema for multiplicada por λ, sem que haja alteração do equilíbrio e composição,
todas aquelas variáveis vêm multiplicadas pelo mesmo factor. Então a relação U = U (S, V, ni)
tem a seguinte propriedade:
U (λ S, λ V, λ ni) = λ U (S, V, ni)
Ou seja, a função U = U (S, V, ni) é homogénea de grau 1. Então podemos escrever a seguinte
relação:
U = S(
∂U
∂U
∂U
) U,V,n j
) V,ni + V ( )S,ni + ∑ n i (
i
∂V
∂S
∂n i
A relação de Euler toma então a forma:
C
U = S T − V p + ∑ niμ i
1
As funções T(S, V, ni), p(S, V, ni) e μi(S, V, ni), são funções homogéneas de grau zero. São
funções independentes da massa (são variáveis intensivas).
Se se conhecerem todas as equações de estado, podemos substituir as variáveis intensivas (como
função das variáveis extensivas) na relação de Euler e recuperar a relação fundamental. Quer
dizer que só a totalidade das equações de estado é que contém toda a informação termodinâmica
sobre o sistema.
Se diferenciarmos a relação de Euler, obtemos:
C
C
1
1
dU = S dT + T dS − V dp - p dV + ∑ n i dμ i + ∑ μ i dn i
Comparando com a relação
C
dU = TdS − pdV + ∑ μ i dn i
i =1
chegamos à conclusão que a seguinte relação é válida:
C
S dT − V dp + ∑ n i dμ i = 0
1
Esta é a relação de Gibbs-Duhem, a qual estabelece uma relação entre as diferenciais das
variáveis intensivas. Isto corresponde a dizer que as variáveis intensivas T, p e μi não são todas
independentes, isto é, não podem todas variar arbitrariamente (uma delas terá uma variação
dependente da variação das outras). O número de parâmetros intensivos capazes de uma variação
70
TERMODINÂMICA APLICADA
2009 Isabel Ambar
independente corresponde ao número de graus de liberdade termodinâmica de um dado sistema.
Um sistema simples com C componentes tem C+1 graus de liberdade.
Se nos referirmos ao esquema da entropia, a relação de Euler e a relação de Gibbs-Duhem terão
as formas seguintes, respectivamente:
S=
C
1
p
μ
U + V − ∑ i ni
T
T
1 T
C
1
p
μ
U d( ) + V d( ) − ∑ n i d( i ) = 0
T
T
T
1
9.3. Potenciais termodinâmicos. Aplicações.
Tanto na representação da energia como na da entropia, os parâmetros extensivos são as variáveis
independentes enquanto os intensivos surgem como derivadas. Ora isto está em desacordo com as
situações práticas, na medida em que, em geral, as variáveis intensivas são muito mais fáceis de
medir e controlar (p. ex., a temperatura) do que as variáveis extensivas (p. ex., a entropia). A
questão que se põe, então, é saber se é possível reformular a teoria de modo a que as variáveis
intensivas passem a ser as variáveis independentes (em vez das extensivas), sem perder a
informação termodinâmica que a relação fundamental fornece. Vamos ver que isso é possível
através da aplicação das transformadas de Legendre. Vejamos como se procede através de um
exemplo.
Para simplificar, considere-se que a relação fundamental corresponde a uma função de uma única
variável Y = Y (X).
Geometricamente, a relação fundamental Y=Y(X) é representada por uma curva no espaço de
coordenadas cartesianas X e Y. A derivada P=dY/dX corresponde ao declive dessa curva.
O objectivo é substituir a variável X como variável independente, pela variável P, em que
P=dY/dX. Isso pode ser feito por eliminação de X entre Y=Y(X) e P=dY/dX=P(X), obtendo-se
Y=Y(P).
Mas se agora quisessemos partir desta última relação (Y função de dY/dX) para voltar a
recuperar a relação fundamental, teríamos de integrar e portanto iríamos obter Y=Y(X) a menos
de uma constante de integração. Quer dizer que alguma da informação originalmente contida na
relação fundamental se teria perdido ao passarmos para a relação Y=Y(P). Do ponto de vista
geométrico, isto quer dizer que o conhecimento de Y em função do declive dY/dX não permite
reconstruir a curva Y=Y(X).
Exemplo
Função Y = X2+3: então P= dY/dX = 2X e portanto X= P/2
P
2
Substituindo vem Y = ( ) 2 + 3 =
P2
+3
4
Para recuperar a relação fundamental original, teríamos de integrar a equação Y −
que levava à relação Y=Y(X) a menos de uma constante de integração.
71
1
(Y')2 = 3 , o
4
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Y
Y
X
X
A solução prática para este problema vem do facto de que uma
dada curva pode ser representada de uma das 2 maneiras: (i)
como o lugar geométrico de pontos (X,Y) que satisfazem a
relação Y=Y(X) ou (ii) através de uma família de linhas
tangentes envolvente da curva. Então, qualquer equação que
nos permita construir a família de tangentes determina bem a
curva.
Y
X
Assim como qualquer ponto no plano é descrito pelos dois
números X e Y, também qualquer linha recta no plano pode
ser descrita pelos dois números P e Ψ (P é o declive e Ψ a
ordenada na origem). Então assim como a relação Y=Y(X)
selecciona um subconjunto de todos os pontos (X, Y)
possíveis, a relação Ψ = Ψ (P) selecciona um subconjunto de
todas as linhas (P, Ψ) possíveis. O conhecimento das
ordenadas na origem (Ψ) das linhas tangentes em função dos
declives (P) permite-nos construir a família das linhas
tangentes e portanto construir a curva de que elas são a
envolvente.
Y
(0, Ψ)
(X,Y)
Ψ
X
Então a relação Ψ = Ψ(P) é totalmente equivalente à relação fundamental (na representação de
Y) Y = Y(X), e portanto pode ser também considerada como uma relação fundamental (agora na
representação de Ψ). Nesta nova relação a variável independente é agora P.
Como calcular esta nova relação a partir de Y=Y(X)? A operação matemática apropriada
denomina-se transformação de Legendre. Vamos ver como obtê-la.
O declive da linha tangente que passa pelo ponto (X, Y) é dado por:
Y−Ψ
P=
X−0
ou
72
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Ψ=Y–PX
Então eliminando X e Y entre esta relação e Y = Y(X) e P=dY/dX=P(X), obtemos Ψ = Ψ (P).
A diferencial de Ψ pode ser obtida a partir daquela relação:
dΨ = dY – P dX – X dP= - X dP
e portanto
dΨ
−X =
dP
Exemplo
Função Y = X2+3
P= dY/dX= 2X e portanto X= P/2. Construindo agora a função Ψ(P):
Ψ(X) = Y – X P = (X2+3) – X (2X) = -X2 + 3
Substituindo X pela função de P (X(P)), obtemos a transformada de Legendre de Y(X):
P
P2
Ψ(P) = −( ) 2 + 3 = −
+3
4
2
Se agora se pretender reconstruir a relação fundamental original:
dΨ
P
e portanto
P = 2X
−X =
=−
dP
2
Como Y = Ψ + X P = −
P2
P
P2
+ 3 + P( ) =
+3
4
4
2
Substituindo P=2X vem: Y = X2 + 3
Resumo
ψ=ψ(P)
- X = dψ/dP
Y=XP+ψ
Eliminando P e ψ vem:
Y = Y(X)
Y=Y(X)
P=dY/dX
ψ=-PX+Y
Eliminando X e Y vem:
ψ = ψ(P)
A generalização da transformação de Legendre a funções com mais de uma variável
independente é simples.
Y = Y (X1, X2, ..., Xn)
Pk =
∂Y
∂X k
Ψ = Y − ∑ X k Pk
k
dΨ = − ∑ X k dPk
k
∂Ψ
∂Pk
Pode haver transformações de Legendre parciais. Nesse caso só se aplica a transformação a parte
das variáveis, considerando-se que as restantes variáveis se mantêm constantes na
transformação.Vejamos então um caso geral de uma relação fundamental da forma:
Y = Y(X1, X2, ..., Xn, Xn+1, …, Xt)
− Xk =
73
TERMODINÂMICA APLICADA
2009 Isabel Ambar
A transformada de Legendre nas variáveis X1, ..., Xn vai ter como variáveis naturais P1, …, Pn,
Xn+1, …, Xt:
Ψ (P1, …, Pn, Xn+1, …, Xt) = Y -
n
∑ Pk X k
k =1
∂Ψ
para k≤n
∂Pk
∂Ψ
para k>n
Pk =
∂X k
− Xk =
n
dΨ = − ∑ X k dPk +
k =1
t
∑
Pk dX k
k = n +1
A aplicação do formalismo anterior à Termodinâmica é evidente. As funções transformadas de
Legendre denominam-se potenciais termodinâmicos. Considere-se então a relação fundamental
na forma:
U = U (S, V, n1, ..., nc)
e vamos então aplicar-lhe transformações de Legendre parciais:
(i) Vamos obter uma função transformada que, em lugar de ser função de S, V, n1, ..., nc, tenha
como variáveis naturais T, V, n1, ..., nc (ou seja, S é substituída por T). Esta transformada
denomina-se energia livre (de Helmholtz) ou potencial de Helmholtz:
F=U−(
∂U
) V,n i S = U − TS
∂S
Então F = F (T, V, n1, ..., nc) e a sua diferencial é:
c
∂F
∂F
∂F
dF = ( )V,n i dT + ( )T, n i dV + ∑ ( )T, V,n j dni = −SdT − pdV + μ1dn1 + ... + μ cdn c
∂T
∂V
i =1 ∂n i
(ii) A função transformada que, em lugar de ser função de S, V, N1, ..., Nc, tem como variáveis
naturais S, p, n1, ..., nc (ou seja, V é substituída por p), denomina-se entalpia:
H = U−(
∂U
)S, n i V = U + pV
∂V
Então H = H (S, p, N1, ..., Nc) e a sua diferencial é:
dH = (
c
∂H
∂H
∂H
) p, n i dS + ( )S, n i dp + ∑ (
)S, p, n j dn i = TdS + Vdp + μ1dn1 + ... + μ c dn c
∂S
∂p
i =1 ∂n i
(iii) A função transformada que, em lugar de ser função de S, V, n1, ..., nc, tem como variáveis
naturais T, p, n1, ..., nc (ou seja, V é substituída por p e S por T), denomina-se potencial de Gibbs
(ou energia livre de Gibbs):
∂U
∂U
G = U − ( ) V, n i S - ( ) S, n i V = U - TS + pV
∂S
∂V
Então G = G (T, p, n1, ..., nc) e a sua diferencial é:
74
TERMODINÂMICA APLICADA
2009 Isabel Ambar
c
∂G
∂G
∂G
dG = ( )p,n i dT + ( )T,n i dp + ∑ ( )T,p,n j dni = -SdT + Vdp + μ1dn1 + ... + μ cdnc
∂T
∂p
i =1 ∂n i
Estas funções transformadas de Legendre são as mais utilizadas. Mas também se podem definir
transformadas de Legendre a partir da relação fundamental na representação da entropia. Estas
funções são designadas por funções de Massieu (Massieu apresentou-as em 1869 precedendo a
apresentação dos potenciais termodinâmicos por Gibbs em 1875).
Exercício sugerido
Defina a função de Massieu cujas coordenadas naturais são 1/T e p/T.
9.4. Relações de Maxwell. Síntese de Max-Born Tisza.
O teorema de Schwartz estabelece a igualdade entre as derivadas parciais mistas de uma função
2
2
contínua. Se f = f(x, y), então ∂ f = ∂ f . Aplicando relações deste tipo às derivadas de 2ª
∂x∂y
∂y∂x
ordem de U(S, V, ni), vamos obter igualdades entre derivadas de variáveis termodinâmicas que
são conhecidas por relações de Maxwell. Por exemplo:
∂2U
∂ 2U
=
∂S∂V ∂V∂S
∂2U
∂2U
=
∂S∂n j ∂n j∂S
2
2
∂ U
∂ U
=
∂V∂n j ∂n j ∂V
→
−(
→
(
→
(
∂p
∂T
) V, n i = ( )S, n i
∂S
∂V
∂μ j
∂S
) V, n i = (
∂μ j
∂V
∂T
)S, V, n i com (ni≠nj)
∂n j
) S,n i = − (
∂p
) S,V, n i com (ni≠nj)
∂n j
Se partirmos da relação fundamental no esquema dos potenciais termodinâmicos vamos obter
novas relações de Maxwell. Por exemplo:
Para F= F (T, V, ni)
dF = - S dT – p dV + ∑μi dni
(
∂S
∂p
) T, n i = ( ) V, n i
∂V
∂T
-(
∂μ j
∂S
) T,V,n i = (
) V,n i
∂n j
∂T
com (ni≠nj)
-(
∂μ j
∂p
) T,V,n i = (
) T,n i
∂n j
∂V
com (ni≠nj)
Existe uma mnemónica que facilita muito a lembrança das relações de Maxwell mais utilizadas.
É designada por síntese de Max Born-Tisza e consiste num quadrado com setas ao longo das
diagonais a apontar para os vértices. Os lados correspondem às funções U, F, G, H e os vértices
adjacentes a cada lado correspondem às variáveis naturais da função associada a esse lado. As
diagonais apontam das variáveis extensivas para as intensivas conjugadas.
75
TERMODINÂMICA APLICADA
2009 Isabel Ambar
V
F
U
S
T
G
H
V
F
T
U
ou
S
-p
G
p
H
A partir desta mnemónica podemos escrever facilmente as diferenciais das funções U, F, H e G.
E podemos obter relações de Maxwell. Por exemplo:
T
V
(
(
∂V
∂T
) p,n i = ( ) S,n i
∂S
∂p
U
∂T
∂p
) S,n i = −( ) V,n i
∂V
∂S
G
S
p
V
T
U
S
p
Em aplicações práticas da Termodinâmica, é frequentemente necessário avaliar uma derivada
parcial e para tal a sua modificação pode ser muito útil. As seguintes identidades são muitas
vezes utilizadas para esse fim:
(
(
∂X
1
) =
∂Y
∂Y Z
( )Z
∂X
∂X
∂X
∂W
)Z = (
)Z (
)Z
∂Y
∂W
∂Y
∂Z
( )X
∂X
( ) Z = − ∂Y
∂Z
∂Y
( )Y
∂X
(relação em cadeia)
(relação cíclica)
76
TERMODINÂMICA APLICADA
2009 Isabel Ambar
(
∂X
∂X
∂X
∂W
)Z = ( )W + (
)Y (
)Z
∂Y
∂Y
∂W
∂Y
9.5. Exemplos de aplicações das relações termodinâmicas
9.5.1. Coeficientes termodinâmicos e calores específicos
Dado que a relação fundamental para um sistema pode vir expressa no esquema de qualquer dos
potenciais termodinâmicos, vamos escolher de entre estes o que tem como coordenadas naturais
duas variáveis que podemos facilmente medir na prática – a temperatura e a pressão – e que é o
potencial de Gibbs (G = G (T, p, n1, ..., nc)). E vamos ver que, a partir das equações de estado
correspondentes a esta relação fundamental, vão surgir derivadas parciais que se identificam com
coeficientes termodinâmicos de utilidade prática.
G = G (T, p, n1, ..., nc)
As derivadas parciais em ordem a T e a p são:
∂G
) T,n i = V(T, p, n 1 , ..., n c )
∂p
∂G
( ) p,n i = − S(T, p, n 1 , ..., n c )
∂T
(
Diferenciemos a primeira equação de estado:
⎛ ∂V ⎞
⎛ ∂V ⎞
⎛ ∂V ⎞
⎟⎟
⎟⎟ dp + ∑ ⎜⎜
dV = ⎜
dn i
⎟ dT + ⎜⎜
∂
n
⎝ ∂T ⎠p, n i
⎝ ∂p ⎠T, n i
i
⎝
⎠p,T, n
j
Ao dividir esta diferencial por V, obtemos a variação relativa do volume como uma soma de
várias contribuições (devidas à variação da temperatura, da pressão e do número de moles dos
componentes):
dV 1 ⎛ ∂V ⎞
1 ⎛ ∂V ⎞
1 ⎛ ∂V ⎞
⎟
⎟⎟ dp + ∑ ⎜⎜
= ⎜
dn i
⎟ dT + ⎜⎜
V V ⎝ ∂T ⎠p, n i
V ⎝ ∂p ⎠T, n i
V ⎝ ∂n i ⎟⎠p,T, n
j
(i) O primeiro termo relaciona-se directamente com o coeficiente de expansão térmica (α):
α=
1 ∂V
( ) p, n i
V ∂T
1
, como facilmente se
T
nR u
1 nR u 1
⎛ ∂V ⎞
=
demonstra: p V = n Ru T e portanto ⎜
. Logo: α =
= .
⎟
p
V p
T
⎝ ∂T ⎠ p, n
No caso de um gás ideal, o coeficiente de expansão térmica é α =
Exercício sugerido:
O efeito do aquecimento global na variação do nível do mar é essencialmente devido à expansão
térmica da água. Faça uma estimativa do valor médio da subida do nível do mar associado ao
aumento de 2ºC da temperatura da camada superior (200 m) do oceano (área da superfície do
oceano ~ 3,6x108 km2; αmed (água do mar) ~ 2281x10-7 ºC-1).
77
TERMODINÂMICA APLICADA
2009 Isabel Ambar
O segundo termo relaciona-se com o coeficiente de compressibilidade isotérmica (βT):
βT = -
1 ∂V
( )T, n
V ∂p i
O sinal negativo que aparece na definição deste coeficiente tem a ver com o facto de um aumento
da pressão levar a uma diminuição do volume e portanto aquela derivada parcial ser negativa mas
nós querermos que o coeficiente seja positivo.
No caso de um gás ideal, o coeficiente de compressibilidade isotérmica é β T =
1
, como se
p
nR u
⎛ ∂V ⎞
nR
1
1
⎟⎟
demonstra: p V = n Ru T e portanto ⎜⎜
. Logo: β T = − (− u ) = .
=−
2
2
V
p
p
⎝ ∂p ⎠ T, n
p
Note-se que este coeficiente é diferente do coeficiente de compressibilidade adiabática (βS)
definido como:
βS = -
1 ∂V
( ) S,n i
V ∂p
De que relação fundamental se deveria ter partido para obter uma equação de estado cuja
diferencial se relaciona directamente com este coeficiente βS? Demonstre que teria de ser da
relação fundamental no esquema da entalpia (H= H(S, p, ni)).
Vamos agora diferenciar a segunda equação de estado:
∂G
( ) p, n i = S(T, p, n1 , ..., n c )
∂T
⎛ ∂S ⎞
⎛ ∂S ⎞
⎛ ∂S ⎞
⎟⎟
dS = ⎜ ⎟ dT + ⎜⎜ ⎟⎟ dp + ∑ ⎜⎜
dn i
⎝ ∂T ⎠p, n i
⎝ ∂p ⎠T, n i
⎝ ∂n i ⎠p,T, n
j
Multipliquemos esta diferencial por T (lembrando que δQ=TdS num processo reversível):
⎛ ∂S ⎞
⎛ ∂S ⎞
⎛ ∂S ⎞
⎟⎟
TdS = T⎜ ⎟ dT + T⎜⎜ ⎟⎟ dp + ∑ T⎜⎜
dn i
⎝ ∂T ⎠p, n i
⎝ ∂p ⎠T, n i
⎝ ∂n i ⎠p,T, n
j
O primeiro termo relaciona-se com a capacidade térmica a pressão constante:
C p = T(
∂S
) p, n
∂T i
Convém lembrar que se falarmos em calor específico (i.e., por unidade de massa), a entropia
∂s
nesta expressão também terá de ser específica: c p = T( ) p, n i
∂T
Também existe a capacidade térmica a volume constante:
C V = T(
∂S
) V, n i
∂T
78
TERMODINÂMICA APLICADA
2009 Isabel Ambar
De que relação fundamental se deveria ter partido para obter uma equação de estado cuja
diferencial se relaciona directamente com este coeficiente Cv? Demonstre que teria de ser da
relação fundamental no esquema da energia livre (F= F(T, V, ni)).
No Anexo I, apresentam-se tabelas que mostram como varia o coeficiente de expansão térmica, o
coeficiente de compressibilidade isotérmica e o calor específico a pressão constante para a água
do mar. Todas estas grandezas dependem da temperatura, da pressão e da salinidade (i.e.,
concentração em sais na água do mar, correspondendo a uma medida do número de moles dos
vários elementos que existem dissolvidos na água do mar). Neste Anexo também se apresentam
tabelas de valores do calor específico de vários gases em função da temperatura.
9.5.2. Relações T dS
A entropia de um sistema fechado pode escrever-se em função de diferentes variáveis (p, V ou T)
e convém escolher as que melhor servem para a resolução de um dado problema (note-se que não
estamos a falar de uma relação fundamental). Vejamos então as 3 opções correspondentes a
escolher como variáveis independentes (T, V), (T, p) ou (p, V).
(i) S = S(T, V)
dS = (
∂S
∂S
) dT + ( ) dV
∂T V
∂V T
C
∂S
) = V
T
∂T V
∂S
∂p
Pelas relações de Maxwell: ( ) = ( )
∂T V
∂V T
Mas, por definição, (
∂V
)
Aplicando a igualdade cíclica a esta derivada parcial: ( ∂p ) = − ∂T p = − α V = α
∂V
∂T V
− βT V βT
( )
∂p T
(
Portanto:
T dS = Cv dT +
αT
dV
βT
Esta relação é útil em processos em que a temperatura e o volume podem variar.
(ii) S = S(T, p)
dS = (
(
∂S
∂S
) dT + ( ) dp
∂T p
∂p T
Cp
∂S
) =
∂T p
T
79
TERMODINÂMICA APLICADA
2009 Isabel Ambar
∂S
∂V
) = −( ) = − αV
∂p T
∂T p
Portanto:
(
T dS = C p dT − α V T dp
Esta relação é útil em processos em que a temperatura e a pressão podem variar.
(iii) S = S(p, V)
dS = (
∂S
∂S
) dp + ( ) dV
∂p V
∂V p
∂V
( )
C
C
C (− β T V) C V β T
∂p T
∂S
∂S
∂T
∂T
( ) = ( ) ( ) = V ( ) = V (−
=
)=− V
∂V
∂p V
∂T V ∂p V
(
T ∂p V
T
T
α V)
Tα
( )
∂T p
(
C p ∂T
Cp
Cp 1
∂S
∂S
∂T
1
) =( ) ( ) =
=
( ) =
∂V p
∂T p ∂V p
T ∂V p
T ∂V
T αV
( )
p
∂T
T dS =
Cp
βT Cv
dp +
dV
α
αV
Esta relação é útil em processos em que a pressão e o volume podem variar.
Vejamos como é possível obter uma relação entre a capacidade térmica a pressão constante
∂S
∂S
( C p = T( ) p, n i ) e a capacidade térmica a volume constante ( C V = T( ) V, n i ) a partir de uma
∂T
∂T
destas relações TdS. Qual devemos escolher? Como na definição dessas grandezas existe uma
derivada parcial de S em ordem a T (com V constante ou com p constante), podemos usar
qualquer uma das relações TdS onde figure dT. Por exemplo:
αT
T dS = Cv dT +
dV
βT
Dividindo por dT e considerando que a pressão se mantém constante:
α T ⎛ ∂V ⎞
αT
⎛ ∂S ⎞
T ⎜ ⎟ = Cv +
αV
⎜
⎟ = Cv +
β T ⎝ ∂T ⎠ p
βT
⎝ ∂T ⎠ p
α2 V T
Cp = Cv +
βT
Esta é a chamada relação de Mayer que exprime a relação entre estas duas capacidades térmicas.
Se nos referirmos a calores específicos (cp e cv), a relação de Mayer terá a forma seguinte (onde v
é o volume específico):
80
TERMODINÂMICA APLICADA
2009 Isabel Ambar
cp = cv +
α2 v T
βT
No caso de um gás ideal (α=1/T, βT=1/p, a relação de Mayer terá a forma:
cp = cv +
R
α2 v T
p
= cv +
v T = cv + u = cv + R
2
βT
M
T
Em termos de calores específicos molares virá:
Cp = Cv +
α 2 v molar T
p
= Cv +
v molar T = C v + R u
βT
T2
9.5.3 Processo de Joule-Thomson (processo de estrangulamento)
Quando nos referimos às máquinas frigoríficas e mencionámos a fase final do ciclo em que havia
expansão adiabática do fluido, com abaixamento da temperatura (de Tq para Tf), dissemos que na
realidade, este processo não é adiabático reversível mas sim isentálpico: o fluido é obrigado a
atravessar uma barreira porosa de uma região de alta pressão para uma de baixa pressão. Esta
queda da pressão leva a um arrefecimento do fluido, sob certas condições. Vamos então estudar
esse processo, o qual é geralmente designado por processo de Joule-Thomson (é um processo de
“throttling” ou de estrangulamento). Considere-se um cilindro termicamente isolado, com 2
êmbolos (isoladores térmicos) em lados opostos de uma parede porosa. Entre um dos lados desta
parede e o êmbolo desse lado está um gás (pressão pi, volume Vi). O outro êmbolo está encostado
à parede porosa evitando a passagem do gás através desta. Os êmbolos são empurrados
simultaneamente, de modo que a pressão pi se mantenha constante enquanto o gás vai passando
através da parede porosa para o outro lado, onde a sua pressão (pf) é menor e também se mantém
constante.
pf
Vf
pi
Vi
Este processo é irreversível porque o gás passa por estados de não-equilíbrio entre o estado inicial
e o estado final, e esses estados não podem ser descritos por variáveis termodinâmicas. Aplicando
a 1ª lei da Termodinâmica a este processo:
ΔU = Q + W, onde ΔU = Uf – Ui
Vf
0
0
Vi
W = − ∫ p f dV − ∫ p i dV = −p f Vf + p i Vi
Então, uma vez que o processo é adiabático (Q = 0) vem: Uf – Ui = - pf Vf + pi Vi
Ui + pi Vi = Uf + pf Vf
ou
81
Hi = Hf
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Neste processo as entalpias inicial e final são iguais mas não podemos dizer que a entalpia se
mantém constante ao longo do processo porque não podemos definir a entalpia em estados de
não-equilíbrio.
Vejamos o que acontece à temperatura no processo de Joule-Thomson em que a variação da
pressão é suficientemente pequena para podermos utilizar análise diferencial.
⎛ ∂H ⎞
⎛ ∂V ⎞
⎟⎟
⎜⎜
+V
− T⎜
⎟
p
∂
⎝ ∂T ⎠p, n
⎝
⎠T, n
⎛ ∂T ⎞
V
dp = −
dp =
dp =
(α T − 1)dp
dT = ⎜⎜ ⎟⎟
⎛ ∂H ⎞
Cp
Cp
⎝ ∂p ⎠H, n
⎜
⎟
⎝ ∂T ⎠p, n
onde se utilizaram as seguintes relações:
dH = T dS + v dp (sendo n=cte.)
⎛ ∂S ⎞
⎛ ∂p ⎞
⎛ ∂S ⎞
⎛ ∂H ⎞
⎛ ∂V ⎞
⎜⎜
⎟⎟
= T⎜⎜ ⎟⎟
+ V⎜⎜ ⎟⎟
= T⎜⎜ ⎟⎟
+ V = - T⎜
+V
⎟
⎝ ∂T ⎠ p, n
⎝ ∂p ⎠ T, n
⎝ ∂p ⎠ T, n
⎝ ∂p ⎠ T, n
⎝ ∂p ⎠ T, n
⎛ ∂p ⎞
⎛ ∂S ⎞
⎛ ∂H ⎞
+ V⎜ ⎟
= Cp
= T⎜ ⎟
⎟
⎜
⎝ ∂T ⎠ p, n
⎝ ∂T ⎠ p, n
⎝ ∂T ⎠ p, n
14243
=0
dT =
V
(α T − 1)dp
Cp
Como no processo de Joule-Thomson, pf < pi (dp<0), então:
Se αT > 1: dT < 0 → há arrefecimento do fluido
Se αT < 1: dT > 0 → há aquecimento do fluido
Portanto αT = 1 corresponde à temperatura de inversão.
No caso de um gás ideal, como α = 1/T vem sempre dT = 0 neste processo. Este resultado não é
de estranhar visto que a entalpia de um gás ideal é função exclusiva da temperatura
(H=U(T)+pV=U(T)+nRu/T)) e portanto não deverá haver variação da temperatura num processo
em que as entalpias inicial e final são iguais.
O coeficiente de Joule-Thomson é definido como:
⎛ ∂T ⎞
V
μ = ⎜⎜ ⎟⎟ =
(α T − 1)
⎝ ∂p ⎠ H C p
Quando o coeficiente de Joule-Thomson é positivo há arrefecimento no processo de
estrangulamento.
Considere-se um espaço T/p e linhas de igual entalpia (isentálpicas) como a figura seguinte
ilustra.
82
TERMODINÂMICA APLICADA
2009 Isabel Ambar
T
Valor máximo da
temperatura de inversão
μ< 0
μ> 0
p
Linha de inversão
(corresponde às temperaturas de inversão, onde μ= 0)
Este diagrama mostra que só pode haver arrefecimento num processo de estrangulamento (μ> 0)
quando a temperatura do fluido for inferior ao valor máximo da temperatura de inversão. Isto
representa um problema para as substâncias para as quais este valor é muito inferior à
temperatura ambiente (p. exº, no caso do hidrogénio, é -68ºC). Neste caso, terá de haver um
arrefecimento prévio até temperaturas abaixo da temperatura máxima de inversão antes de se
continuar o arrefecimento pelo processo de estrangulamento.
Considerando os gases reais a obedecerem à equação de estado de van der Waals, pode-se
mostrar (Callen, 1985) que a temperatura de inversão é dada pela expressão aproximada:
2a
Tinv =
b Ru
onde a e b são as constantes de van der Waals para o gás. Usando aquela expressão aproximada:
Gás
Ne
H2
N2
O2
CO2
a (Pa m6)
0,0215
0,0248
0,136
0,138
0,401
b (10-6 m3)
17,1
26,6
38,5
32,6
42,7
Tinv (K)
302
224
850
1020
2260
Na realidade, a temperatura de inversão depende da pressão e portanto os valores reais diferem
um pouco dos que são dados por esta tabela.
83
TERMODINÂMICA APLICADA
2009 Isabel Ambar
10. Equilíbrio de sistemas termodinâmicos.
10.1. Princípio do mínimo da energia
O princípio do máximo da entropia caracteriza o estado de equilíbrio como tendo um máximo de
entropia para uma dada energia total. Agora vamos ver que um estado de equilíbrio também pode
ser caracterizado por um mínimo de energia para uma dada entropia total.
Demonstração
Queremos mostrar que se a energia não for mínima a entropia não pode ser máxima no equilíbrio
e inversamente.
(i) Argumento físico
Vamos supôr que o sistema está em equilíbrio mas a energia não tem o seu valor menor possível
consistente com a entropia dada. Então podíamos retirar energia do sistema (sob a forma de
trabalho) mantendo S constante, e depois devolver a mesma quantidade de energia ao sistema
mas agora na forma de calor. Então a entropia do sistema aumentaria (đQ = T dS) e o sistema
teria voltado ao valor inicial de energia mas agora com uma entropia maior. Isto é inconsistente
com o facto de que o estado inicial era o estado de entropia máxima. Conclusão: o equilíbrio
original devia corresponder a um mínimo de energia compatível com o valor da sua entropia.
(ii) Argumento matemático
O máximo da entropia (sendo S = S(U, X1, …, Xn)) corresponde a:
(
∂S
=0
)
∂X i U, X j
e
∂S
)
∂Xi U, X j
∂U
∂S
Pi = (
)S, X = −
)
=0
= −T(
S
∂
∂Xi
∂Xi U, X j
j
( )X , X
∂U i j
(
∂ 2S
) U, X < 0
j
∂X i 2
(
Portanto ( ∂U )
o que quer dizer que U é um extremo. Vejamos se é um máximo (2ª
=0
∂X i S, X j
derivada negativa), um mínimo (2ª derivada positiva) ou um ponto de inflexão (2ª derivada nula).
(
∂P
∂P
∂P
∂2U
∂U
) S, X = ( i ) S, X = ( i ) U, X + ( i ) X , X (
)
=
2
X
∂X i
∂ i
∂U i j ∂X i S, X j
j
j
j
∂X i
142
4 43
4
Pi
se Pi = 0, então:
84
TERMODINÂMICA APLICADA
2009 Isabel Ambar
⎤
⎡ ⎛ ∂S ⎞
⎟
⎥
⎢ ⎜
∂ 2S
∂ 2S
(
) U, X
(
)
⎥
⎢ ⎜⎝ ∂Xi ⎟⎠
2
U, X j ⎥
j ⎛ ∂S ⎞
∂Xi
∂Pi
∂Xi∂U U, X j
∂ ⎢
∂ 2S
⎟
=−
=(
+⎜
−
)
= −T(
>0
) U, X =
⎥
⎢
⎜ ∂X ⎟
2 U, X j
2
∂Xi
⎛ ∂S ⎞
j ∂Xi ⎢ ⎛ ∂S ⎞
X
∂
∂
S
⎛
⎞
i
⎝
⎠
⎥
⎜
⎟
⎜
⎟
U, X j ⎜
i
⎟
⎝ ∂U ⎠X , X
⎢ ⎝ ∂U ⎠ X , X ⎥
142
4 43
4 ⎝ ∂U ⎠ Xi , X j
i
j
i
j
⎥
⎢
=0
⎣
⎦
ou seja, U é um mínimo.
O facto de a mesma situação (equilíbrio de um sistema) poder ser descrita pelos dois critérios de
extremo (S máxima para uma dada energia ou U mínima para uma dada entropia) é análogo à
definição de círculo em geometria: um círculo pode ser caracterizado como uma figura
geométrica bidimensional de área máxima para um dado perímetro ou, alternativamente, como
uma figura geométrica bidimensional de perímetro mínimo para uma dada área.
10.2. Condições de equilíbrio em termos dos potenciais termodinâmicos.
Como vimos, as transformadas de Legendre permitem exprimir as relações fundamentais em
função de conjuntos de variáveis independentes que podem ser escolhidas consoante a
conveniência de um dado problema. Mas esta vantagem seria inútil se não houvesse um princípio
de extremo aplicável nessas representações.
Vamos começar por considerar um sistema composto por dois subsistemas em contacto com um
reservatório de temperatura. Vamos remover alguns constrangimentos e procurar as condições de
equilíbrio, começando por aplicar o princípio do mínimo da energia (isto é, a condição de a
energia ser mínima para um dado valor da entropia):
d(U + UR) = 0
(1)
(2)
d2(U + UR) >0
com a condição d(S+SR) = 0
Reservatório TR
As condições impostas dependem dos constrangimentos internos do sistema composto. Vamos
considerar dois casos: (i) parede interna móvel e impermeável e (ii) parede interna rígida e
permeável ao componente k.
(i) Parede móvel e impermeável
dNj(1) =
d(V(1) + V(2)) = 0
dNj(2) = 0 (para qualquer j)
d(U+UR) = dU(1) + dU(2) + dUR = T(1) dS(1) - p(1) dV(1) + T(2) dS(2) – p(2) dV(2)+TR dSR
Mas dSR = - dS = - d(S(1) + S(2))
Então:
d(U+UR) = (T(1) - TR) dS(1) + (T(2) - TR) dS(2)+ (p(1) - p(2)) dV(2) = 0
T(1) = TR
T(2) = TR
p(1) = p(2)
85
TERMODINÂMICA APLICADA
2009 Isabel Ambar
(ii) Parede rígida e permeável ao componente k
dV(1) = dV(2) = 0
dNj(1) = dNj(2) = 0
d(Nk(1) + Nk(2)) = 0
d(U+UR) = dU(1) + dU(2) + dUR = T(1) dS(1) + μk(1) dNk(1) + T(2) dS(2) + μk(2) dNk(2)
+ TR dSR
Mas dSR = - dS = - d(S(1) + S(2))
Então:
d(U+UR) = (T(1) - TR) dS(1) + (T(2) - TR) dS(2)+ (μk(1) - μk(2)) dNk(1) = 0
T(1) = TR
T(2) = TR
μk(1) = μk(2)
Em qualquer dos casos o reservatório mantém a temperatura do sistema constante. Podemos
escrever então a relação d(U+UR) = 0 na forma:
d(U+UR) = dU + TR dSR = dU - TR dS = d(U - TR S) = 0
(visto que TR é constante)
E a relação d2(U+UR) > 0 na forma:
d2(U+UR) = d2U + d (TR dSR) = d2U - TR d2S = d2 (U - TR S) > 0
R
(visto que T é constante e dSR = -dS)
Então a grandeza (U- TR S) é mínima no estado de equilíbrio. Como a temperatura de cada um
dos subsistemas, e portanto do sistema composto, é igual à temperatura do reservatório no estado
de equilíbrio, então (U- T S) = (U- TR S), e portanto dF = 0 e d2F>0 sujeito à condição T = TR.
Princípio do mínimo do potencial de Helmholtz (energia livre)
O valor de equilíbrio de qualquer parâmetro interno sem constrangimentos, num sistema em
contacto diatérmico com um reservatório de calor, minimiza a energia livre de entre o conjunto de
estados para os quais T = TR.
Se o sistema estivesse em contacto com um reservatório de pressão:
d(U+UR) = dU - pR dVR = dU + p dV = d(U + p V) = 0
d2(U+UR) = d2(U + pR V) = d2(U + p V) = d2H > 0
Princípio do mínimo da entalpia
O valor de equilíbrio de qualquer parâmetro interno sem constrangimentos, num sistema em
contacto com um reservatório de pressão, minimiza a entalpia de entre o conjunto de estados para
os quais p = pR.
86
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Princípio do mínimo do potencial de Gibbs
O valor de equilíbrio de qualquer parâmetro interno sem constrangimentos, num sistema em
contacto com um reservatório de calor e de pressão, minimiza o potencial de Gibbs de entre o
conjunto de estados para os quais T = TR e p = pR (temperatura e pressão constantes e iguais às
dos respectivos reservatórios).
Podemos generalizar então:
O valor de equilíbrio de qualquer parâmetro interno sem constrangimentos num sistema em
contacto com um conjunto de reservatórios com parâmetros intensivos P1R, P2R,... minimiza a
transformada de Legendre cujas coordenadas naturais são P1, P2,..., de entre o conjunto de estados
para os quais P1 = P1R, P2= P2R, …
Na prática, muitos dos processo são realizados em recipientes rígidos mas com paredes
diatérmicas, de modo que a atmosfera ambiente actua como um reservatório de calor. Nesse caso,
a representação mais conveniente é a que utiliza a energia livre F = F (T, V, N1, N2, ...).
11. Propriedades físicas de uma substância pura. Transições de fase.
11.1. Postulado de estado
Um objectivo importante da Termodinâmica é determinar relações funcionais entre certas
propriedades macroscópicas de um sistema em equilíbrio. Para tal, é necessário conhecer o
número de variáveis independentes necessário para definir o estado do sistema em dadas
condições. Uma classe de propriedades importante é a das propriedades termostáticas ou
intrínsecas, que podem ser avaliadas por observações da substância que compõe o sistema sem
referência às vizinhanças do sistema. Como exemplos, temos a pressão, a temperatura, a
densidade, a energia interna. Como qualquer destas propriedades caracteriza uma média de algum
tipo de comportamento das partículas que compõem o sistema, é natural que elas estejam
relacionadas entre si. Com o objectivo de desenvolver relações entre propriedades macroscópicas
intrínsecas, é necessário saber quantas dessas propriedades podem variar independentemente.
A experiência mostrou que o número de variáveis independentes depende da natureza da situação
física. Com base em dados experimentais para substâncias homogéneas (com uma única fase),
chegou-se à seguinte afirmação, conhecida como o postulado de estado:
O número de propriedades intrínsecas independentes necessárias para determinar o estado de uma
substância é igual ao número de modos de trabalho quase-estático relevantes (i.e., que podem
eventualmente alterar o estado da substância durante um processo) mais um.
No caso de sistemas simples, em que só um modo de trabalho quase-estático pode afectar o
sistema, o postulado de estado diz-nos:
O estado de equilíbrio de uma substância simples, homogénea, é determinado ao especificar-se
os valores de quaisquer duas propriedades intrínsecas independentes.
Os estados de equilíbrio de uma substância simples e homogénea podem ser representados
através de uma superfície no espaço em que as coordenadas são propriedades intrínsecas
87
TERMODINÂMICA APLICADA
2009 Isabel Ambar
relevantes para esse sistema simples. Apesar de podermos escolher quaisquer propriedades
intrínsecas, é conveniente optar por propriedades facilmente mensuráveis e que tenham um
significado físico simples. Esta razão leva-nos a escolher a pressão, a temperatura e o volume
específico.
Vamos considerar substâncias puras, isto é, substâncias com uma composição química
homogénea e invariável independentemente da fase em que estão. Por exemplo, água líquida,
mistura de água líquida e vapor de água, vapor de água, mistura de gelo e água líquida
correspondem todas a substâncias puras. Por vezes, a mistura de gases, tal como o ar, é
considerada uma substância pura desde que não haja mudança de estado.
11.2. Processos de transição de fase de substâncias puras
Durante o processo de transição de fase de uma substância, há coexistência das duas fases.
Um líquido (vapor) que não está prestes a vaporizar (condensar) denomina-se líquido
comprimido (vapor sobreaquecido). Um líquido (vapor) que está prestes a vaporizar denomina-se
líquido saturado (vapor saturado). A figura seguinte representa, num diagrama temperaturavolume, o processo de aquecimento de água a uma dada pressão.
T
Líquido
saturado
Líquido
comprimido
Mistura
saturada
líquido-vapor
Vapor
sobreaquecido
Vapor
saturado
v
Como se sabe, a água ferve a 100ºC mas só se a pressão for de 1 atmosfera (1 atm=101,325 kPa).
Se a pressão for maior, a água entra em ebulição a uma temperatura superior (p. exº, a 500 kPa, a
água entra em ebulição a 151,8ºC). A tensão do vapor aumenta com a temperatura do líquido
porque quanto maior for a energia cinética das moléculas (que depende da temperatura) mais
moléculas conseguem escapar para a fase de vapor. Se o líquido estiver em contacto com a
atmosfera, quando a tensão do vapor igualar a pressão atmosférica, o líquido entra em ebulição (a
temperatura à qual a tensão do vapor é igual à pressão atmosférica é a temperatura de ebulição).
A uma dada pressão, a temperatura à qual uma substância pura muda de fase denomina-se
temperatura de saturação (Tsat). Do mesmo modo, a uma dada temperatura, a pressão à qual uma
substância pura muda de fase denomina-se pressão de saturação (psat). Há tabelas de pressão de
saturação para diferentes temperaturas (ou vice-versa) disponíveis para quase todas as
substâncias.
A tabela seguinte mostra alguns valores de psat para o caso da água pura. Esta tabela indica, por
exemplo, que para a água mudar de fase (ebulição ou condensação) a 25ºC a pressão tem de ser
88
TERMODINÂMICA APLICADA
2009 Isabel Ambar
3,17 kPa e que para mudar de fase a 250ºC a pressão tem de ser 3976 kPa. Também indica que
pode congelar baixando a pressão para valores inferiores a 0,61 kPa.
Temperatura (ºC)
-10
-5
0
5
10
15
20
25
30
40
50
100
150
200
250
300
Pressão de saturação (kPa)
0,26
0,40
0,61
0,87
1,23
1,71
2,34
3,17
4,25
7,39
12,35
101,4 (1 atm)
476,2
1555
3976
8588
Quando temos água em contacto com o ar, há tendência para a água se evaporar de modo a ser
atingido o equilíbrio entre a água na fase líquida e a água na fase de vapor que existe no ar
circundante. Esse equilíbrio entre fases é atingido quando a pressão do vapor no ar for igual à
pressão de saturação da água à temperatura a que está. A humidade relativa do ar é a razão entre a
pressão de vapor no ar a uma dada temperatura e o valor máximo da pressão de vapor (pressão de
saturação) para essa temperatura.
Apesar dos processos de evaporação e de ebulição corresponderem à mudança de fase de líquido
para vapor, há diferenças entre eles. A evaporação é um fenómeno de superfície que ocorre na
interface líquido-vapor quando a pressão do vapor é menor do que a pressão de saturação do
líquido à temperatura a que está (exemplos: evaporação da água da superfície do oceano,
evaporação da água nas roupas que estão a secar ao ar, evaporação do suor do corpo humano,
etc.).
A ebulição ocorre em todo o corpo do líquido, começando geralmente na interface sólido-líquido
quando um líquido está em contacto com uma superfície a uma temperatura suficientemente
acima da temperatura de saturação do líquido (p. exº, à pressão atmosférica, a água líquida em
contacto com uma superfície sólida a 110ºC entra em ebulição porque a temperatura de saturação
é 100ºC a 1 atm). No processo desprendem-se bolhas de vapor junto à superfície quente, as quais
sobem até à superfície livre do líquido.
89
TERMODINÂMICA APLICADA
2009 Isabel Ambar
A quantidade de energia absorvida ou libertada durante uma transição de fase denomina-se calor
latente - calor latente de fusão (Lf, transição fase sólida-fase líquida), calor latente de vaporização
(Lv, transição fase líquida-fase gasosa). Os valores dos calores latentes de uma dada substância
dependem da temperatura ou da pressão à qual se dá a transição de fase (no caso da água à
pressão de 1 atm, Lf = 333,7 kJ/kg, Lv = 2256,5 kJ/kg).
Durante o processo de transição de fase, há uma
relação entre a pressão e a temperatura:
Tsat = f(psat). A curva correspondente a esta
relação é chamada curva de saturação. Na figura
está representada a curva de saturação líquidovapor para uma substância pura, onde se vê que a
temperatura de saturação aumenta com a pressão.
psat
Tsat
Uma aplicação prática deste facto é a panela de pressão, a qual, por aumentar a pressão a que está
submetida a água com os alimentos, leva a que estes sejam cozidos a uma temperatura superior à
que se consegue com uma panela normal (por exemplo, à pressão de 3 atm a temperatura de
ebulição é 134ºC). Uma consequência da relação Tsat = f(psat) é a temperatura de ebulição da água
ser menor numa região de grande altitude do que ao nível do mar (p. exº, a 2000 m de altitude a
temperatura de ebulição é 93,3ºC, o que corresponde à pressão de 79,5 kPa). As autoclaves,
semelhantes a panelas de pressão, são utilizadas para esterilizar instrumentos de cirurgia (em
geral, atingem temperaturas de cerca de 120ºC e pressões de cerca de 2 atm).
A temperatura de vaporização do azoto à pressão atmosférica é -196ºC. Consequentemente, para
haver azoto líquido à pressão atmosférica, ele terá de estar a -196ºC. Ele ficará a esta temperatura
até passar todo à fase gasosa. Por esta razão, o azoto líquido é utilizado em muitas aplicações
práticas: banho líquido a baixa temperatura, queimaduras localizadas da pele (para tirar sinais,
verrugas, etc.)
O processo de arrefecimento por vácuo é utilizado para a conservação de vegetais com folhas,
tais como os espinafres. O processo consiste na redução da pressão de uma câmara selada até ser
atingida a pressão de saturação correspondente à temperatura final pretendida. O calor de
vaporização durante o processo é retirado dos vegetais.
11.3. Diagramas de propriedades para processos de transição de fase
11.3.1. Diagrama T-v
As mudanças de fase da água para diferentes valores da pressão podem ser representadas num
diagrama T-v (sendo v o volume específico).
90
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Adaptado de Y. A. Çengel & M. A. Boles, 2006
(1 MPa=1000 kPa)
Se a pressão for superior à pressão atmosférica, há diferenças relativamente ao processo de
aquecimento da água:
- a água entra em ebulição a uma temperatura superior
- o volume específico do líquido saturado é maior
- o volume específico do vapor saturado é menor
Em consequência, a linha entre os estados de líquido e de vapor saturados é mais curta. À medida
que a pressão a que se dá o processo vai aumentando, esta linha vai ficando cada vez mais curta
até que fica reduzida a um ponto quando a pressão atinge 22,06 MPa no caso da água. Este ponto
denomina-se ponto crítico e corresponde ao estado em que o líquido saturado e o vapor saturado
são idênticos. Para pressões acima da pressão crítica não há um processo de mudança de fase, há
só uma fase.
A tabela seguinte dá os valores da pressão, temperatura e volume específico no ponto crítico para
algumas substâncias.
-1
Substância
Massa
molar (kg
-1
kmol )
Tcrítica (K)
pcrítica (MPa)
vcrítico (m3 kmol )
Ar
Dióxido de carbono
Hélio
Azoto
Água
28,97
44,01
4,003
28,013
18,015
132,5
304,2
5,3
126,2
647,1
3,77
7,39
0,23
3,39
22,06
0,0883
0,0943
0,0578
0,0899
0,0560
Exercício: Apresente os valores críticos para a água nas seguintes unidades: ºC, kPa e m3 kg-1.
(373,95ºC, 22,06x103kPa, 0,003106 m3 kg-1)
A linha que une os estados de líquido saturado (vapor saturado) denomina-se linha de líquido
saturado (linha de vapor saturado). Estas linhas encontram-se no ponto crítico, formando um
domo. O interior desta linha é a região de mistura de líquido-vapor saturados onde coexistem
91
TERMODINÂMICA APLICADA
2009 Isabel Ambar
duas fases. No exterior do domo, do lado esquerdo temos a região de líquido comprimido e do
lado direito a região de vapor sobreaquecido.
Adaptado de Y. A. Çengel & M. A. Boles, 2006
11.3.2. Diagrama p-v
No caso de um diagrama p-v para uma substância pura, a forma da curva que encerra a região de
saturação líquido-vapor é muito semelhante à do diagrama T-v, mas as linhas de igual
temperatura são descendentes para v crescente (enquanto as linhas de pressão constante no
diagrama T-v eram ascendentes com v).
Adaptado de Y. A. Çengel & M. A. Boles, 2006
11.3.3. Diagrama p-v incluindo a fase sólida
Vamos agora incluir também no diagrama a fase sólida e as regiões de transição sólida-líquida e
sólida-vapor. A maior parte das substâncias contraem-se durante o processo de solidificação mas
há algumas, como a água, que se expandem ao solidificar. Os respectivos diagramas p-v estão
representados nas figuras seguintes:
92
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Diagrama p-v para uma substância que se contrai ao solidificar
Adaptado de Y. A. Çengel & M. A. Boles, 2006
Diagrama p-v para uma substância que se expande ao solidificar (p. exº, a água)
Adaptado de Y. A. Çengel & M. A. Boles, 2006
Em determinadas circunstâncias, as três fases (sólida, líquida e gasosa) de uma substância pura
coexistem em equilíbrio. Nos diagramas p-v ou T-v, esses estados triplos formam uma linha
denominada linha tripla. Esses estados têm a mesma pressão e temperatura mas diferentes
volumes específicos. Num diagrama p-T, essa linha aparece como um ponto – o ponto triplo. No
caso da água, o ponto triplo é caracterizado por uma pressão de 0,61 kPa e uma temperatura de
0,01ºC.
Substância
Massa molar
(kg kmol-1)
Ttriplo (K)
Dióxido de carbono
Hélio
Azoto
Água
44,01
4,003
28,013
18,015
216,55
2,19
63,18
273,16
93
ptriplo (kPa)
517
5,1
12,6
0,61
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Como o ponto triplo para cada substância corresponde a uma só temperatura e pressão, é um
excelente estado de referência para o estabelecimento de uma escala de temperaturas (como se
referiu no ponto 2.2 deste texto).
Nenhuma substância pode existir no estado líquido estável a pressões abaixo da pressão do ponto
triplo.
No caso de substâncias que se contraem ao solidificar, nenhuma substância pode existir no estado
líquido estável a temperaturas abaixo da temperatura do ponto triplo. Contudo, no caso de
substâncias que se expandem ao solidificar (como a água), podem existir na fase líquida a altas
pressões com temperaturas abaixo do ponto triplo. Por exemplo, a água não pode existir na forma
líquida em equilíbrio à pressão atmosférica com temperaturas abaixo dos 0ºC mas pode existir na
fase líquida a -20ºC à pressão de 200 MPa. Estas conclusões são mais fáceis de apreender
examinando o diagrama p-T da secção seguinte.
Há dois modos para uma substância passar de sólido a vapor: (i) passando primeiro à fase líquida
e depois desta à fase de vapor; (ii) passando directamente à fase de vapor se for a pressões abaixo
do ponto triplo. Esta mudança de fase denomina-se sublimação. Para substâncias que têm o ponto
triplo a uma pressão acima da pressão atmosférica, como o CO2, a sublimação é a única forma de
passar da fase sólida (neve carbónica no caso do CO2), para a fase gasosa à pressão atmosférica.
11.3.4. Diagrama p-T
O diagrama p-T é denominado frequentemente como diagrama de fase porque as 3 fases estão
separadas umas das outras por 3 linhas – de sublimação, de fusão e de vaporização. Estas linhas
convergem no ponto triplo e a linha de vaporização termina no ponto crítico.
Adaptado de Y. A. Çengel & M. A. Boles, 2006
A partir dos diagramas que vimos até agora podemos concluir que, para um sistema com um
único componente: (i) quando há uma única fase temos duas variáveis independentes (T,v ou p,v
ou p,T), (ii) quando há duas fases em equilíbrio, só temos uma variável independente e (iii)
94
TERMODINÂMICA APLICADA
2009 Isabel Ambar
quando temos três fases em equilíbrio (ponto triplo) nenhuma das variáveis é independente. Estas
relações podem ser resumidas na lei das fases de Gibbs:
f+L=c+2
sendo f o número de fases em coexistência, L o número de graus de liberdade (i.e., o número de
parâmetros intensivos capazes de variação independente), c o número de componentes.
11.3.5. Superfície p-v-T
Já vimos que o estado de uma substância simples é fixado por quaisquer duas propriedades
intensivas independentes. Uma vez fixadas essas duas propriedades independentes, quaisquer
outras propriedades tornam-se dependentes. Podemos então representar o comportamento de uma
substância no espaço p-v-T como uma superfície cujos pontos representam estados de equilíbrio.
Os diagramas bidimensionais a que nos referimos anteriormente não são mais do que projecções
desta superfície p-v-T nos planos apropriados.
Diagrama p-v-T para uma substância que se contrai ao solidificar
Adaptado de Y. A. Çengel & M. A. Boles, 2006
Diagrama p-v-T para uma substância que se expande ao solidificar
Adaptado de Y. A. Çengel & M. A. Boles, 2006
95
TERMODINÂMICA APLICADA
2009 Isabel Ambar
11.4. Tabelas de propriedades. Aplicações.
Para a maior parte das substâncias, a relação entre propriedades termodinâmicas é demasiado
complexa para ser expressa por simples equações. São então utilizadas tabelas que resultam de
medições e de cálculos utilizando fórmulas que relacionam propriedades termodinâmicas não
mensuráveis com outras que são mensuráveis.
Os exemplos que se seguem correspondem a tabelas de propriedades termodinâmicas para a água
no estado de vapor saturado e líquido saturado e no estado de vapor sobreaquecido.
Antes de ver exemplos de aplicação dessas tabelas, vamos deduzir algumas relações
termodinâmicas que são utilizadas para calcular a variação da entalpia específica, da energia
interna específica e da entropia específica de uma substância pura durante uma mudança de
estado (não esquecendo que, no caso de uma substância pura, só precisamos de 2 variáveis
independentes para especificar o estado).
11.4.1. Relações termodinâmicas envolvendo entalpia, energia interna e entropia
(i) Entalpia específica em função de duas variáveis independentes - p e T
h = h (T, p)
dh = (
∂h
∂h
) p dT + ( ) T dp
∂T
∂p
Mas dh = T ds + v dp e portanto:
96
TERMODINÂMICA APLICADA
2009 Isabel Ambar
⎛ ∂s ⎞
⎛ ∂h ⎞
⎜ ⎟ = T⎜ ⎟ = c p
⎝ ∂T ⎠ p
⎝ ∂T ⎠ p
⎛ ∂h ⎞
⎛ ∂s ⎞
⎛ ∂v ⎞
⎜⎜ ⎟⎟ = v + T⎜⎜ ⎟⎟ = v − T⎜ ⎟
⎝ ∂T ⎠ p
⎝ ∂p ⎠ T
⎝ ∂p ⎠ T
Então:
⎡
⎛ ∂v ⎞ ⎤
dh = c p dT + ⎢ v − T⎜ ⎟ ⎥ dp
⎝ ∂T ⎠ p ⎥⎦
⎢⎣
dh p = c p dTp
Ao longo de uma isóbara:
⎡
⎛ ∂v ⎞ ⎤
dh T = ⎢ v − T⎜ ⎟ ⎥ dp T
⎝ ∂T ⎠ p ⎥⎦
⎢⎣
A variação da entalpia específica na passagem de um estado (T1, p1) para um estado (T2, p2),
pode ser obtida por integração daquela expressão geral de dh:
Ao longo de uma isotérmica:
T2
p2 ⎡
⎛ ∂v ⎞ ⎤
⎢
v
T
−
∫ ⎢ ⎜⎝ ∂T ⎟⎠ p ⎥⎥ dp
⎦
T1
p1 ⎣
Para integrar o 1º termo, é necessário conhecer o calor específico a pressão constante para uma
dada pressão. Para integrar o 2º termo, é necessário conhecer uma equação de estado que dê a
relação entre p, v e T.
h 2 − h1 =
∫ c p dT +
(ii) Energia interna específica em função de duas variáveis independentes - T e v
u = u(T, v)
∂u
∂u
du = ( ) v dT + ( ) T dv
∂T
∂v
Mas du = T ds - p dv
⎛ ∂s ⎞
⎛ ∂u ⎞
⎜ ⎟ = T ⎜ ⎟ = cv
⎝ ∂T ⎠ v
⎝ ∂T ⎠ v
⎛ ∂p ⎞
⎛ ∂u ⎞
⎛ ∂s ⎞
⎜ ⎟ = T⎜ ⎟ − p = T⎜ ⎟ − p
⎝ ∂v ⎠ T
⎝ ∂v ⎠ T
⎝ ∂T ⎠ v
Então:
Ao longo de uma isocórica:
⎤
⎡ ⎛ ∂p ⎞
du = c v dT + ⎢T⎜ ⎟ − p ⎥ dv
⎦
⎣ ⎝ ∂T ⎠ v
du v = c v dTv
⎡ ⎛ ∂p ⎞
⎤
du T = ⎢T⎜ ⎟ − p⎥ dv T
⎣ ⎝ ∂T ⎠ v
⎦
Para o cálculo da variação da energia específica na passagem de um estado (T1, v1) para um
estado (T2, v2), o procedimento seria semelhante ao que já vimos para a entalpia.
Ao longo de uma isotérmica:
97
TERMODINÂMICA APLICADA
2009 Isabel Ambar
T2
v2
⎡ ⎛ ∂p ⎞
⎤
c
dT
+
∫ v
∫ ⎢⎣T⎜⎝ ∂T ⎟⎠ v − p⎥⎦ dv
T1
v1
É claro que se conhecermos (h2 – h1) podemos calcular (u2 - u1), ou vice-versa, a partir da
relação:
u2 – u1 = h2 – h1 – (p2v2 – p1v1)
u 2 − u1 =
Exercício
Determinar expressões semelhantes para a variação da entropia específica, utilizando as relações
funcionais s = s(T, p) e s= s(T, v)
T2
p2
T2
v2
dT
⎛ ∂v ⎞
s 2 − s1 = ∫ cp
− ∫ ⎜ ⎟ dp
T p1⎝ ∂T ⎠p
T1
dT
⎛ ∂p ⎞
s 2 − s1 = ∫ c v
+ ∫ ⎜ ⎟ dv
T v1⎝ ∂T ⎠ v
T1
(iii) Equação de Clapeyron
A equação de Clapeyron é um bom exemplo de como a variação de uma propriedade que não é
mensurável directamente (por exemplo, a entalpia), pode ser determinada a partir de medições de
pressão, temperatura e volume específico.
Durante um processo de transição de fase, a pressão é a pressão de saturação, a qual depende
apenas da temperatura e é independente do volume específico. Então a derivada parcial
⎛ dp ⎞
⎛ ∂p ⎞
⎜ ⎟ pode ser expressa como uma derivada total ⎜
⎟ que corresponde ao declive da curva
⎝ ∂T ⎠ v
⎝ dT ⎠ sat
de saturação num diagrama p-T, para um dado estado de saturação. Este declive é independente
do volume específico.
⎛ ∂s ⎞
⎛ ∂s ⎞
⎟ dv + ⎜
⎟ dT
⎝ ∂v ⎠ T
⎝ ∂T ⎠ v
Considerando s=s(v, T), vem: ds = ⎜
Como durante uma transição de fase, T se mantém constante, então:
⎛ ∂s ⎞
ds = ⎜ ⎟ dv
⎝ ∂v ⎠ T
⎛ ∂p ⎞
⎛ ∂s ⎞
⎟ = ⎜ ⎟ , vem:
⎝ ∂v ⎠ T ⎝ ∂T ⎠ v
Aplicando a seguinte relação de Maxwell ⎜
⎛ ∂p ⎞
⎛ dp ⎞
ds = ⎜ ⎟ dv = ⎜
⎟ dv
⎝ ∂T ⎠ v
⎝ dT ⎠ sat
Podemos então integrar esta expressão entre dois estados saturados à mesma temperatura.
Por exemplo, para uma mudança de fase de líquido (f) para vapor (g), a integração de ds dará:
⎛ dp ⎞
sg − sf = ⎜
⎟ (v g − v f )
⎝ dT ⎠ sat
Como durante o processo, a pressão também se mantém constante, dh = T ds + v dp ≡ T ds e
portanto:
98
TERMODINÂMICA APLICADA
2009 Isabel Ambar
g
g
g
f
f
f
∫ dh = h g − h f = ∫ T ds = T ∫ ds = T (s g − s f )
⎛ dp ⎞
⎟ (v g − v f )
⎝ dT ⎠ sat
hg − hf
h fg
L vap
⎛ dp ⎞
=
=
⎜ ⎟ =
⎝ dT ⎠ sat T (v g − v f ) T v fg T v fg
Então h g − h f = T (s g − s f ) = T ⎜
Esta é a equação de Clapeyron (Clapeyron, físico, 1799-1864), a qual permite determinar a
entalpia de vaporização (hfg), ou seja, o calor latente (Lvap), a uma dada temperatura, por medição
do declive da curva de saturação, num diagrama p-T, conhecendo os volumes específicos das
duas fases saturadas a essa temperatura.
No caso em que o volume específico da fase líquida é muito menor que o da fase gasosa (vf<<vg),
podemos desprezar vf. Se utilizarmos a equação de estado dos gases ideais, podemos substituir vg
L vap p
⎛ dp ⎞
⎟ = 2
⎝ dT ⎠ sat
T R
por RT/p e obter a seguinte expressão aproximada: ⎜
A equação de Clapeyron é aplicável a qualquer transição de fase e portanto podemos generalizar
(fases 1 e 2):
h
⎛ dp ⎞
⎜ ⎟ = 12
⎝ dT ⎠ sat T v12
Exercício: Utilize a equação de Clapeyron para estimar o valor da entalpia de vaporização da
água à temperatura de 20ºC. Compare com o valor dado pelas tabelas.
11.4.2. Desenvolvimento de tabelas de propriedades termodinâmicas
Há muitos modos de gerar tabelas de propriedades termodinâmicas a partir de dados
experimentais. Vamos dar exemplos considerando apenas as fases líquida e de vapor.
Vamos supor que foram obtidos em laboratório, os seguintes dados para uma substância pura: (i)
pressões e temperaturas do vapor saturado; (ii) pressão, temperatura e volume específico na
região de vapor; (iii) densidade do líquido saturado e pressão e temperatura críticas; (iv) calor
específico do vapor.
Os passos serão os seguintes:
- Determinar a equação para a curva de saturação de vapor que melhor se ajusta aos dados. Por
B
exemplo: ln p sat = A + + C ln T + D T
T
Com esta equação, pode-se obter a pressão de saturação para qualquer temperatura.
- Determinar a equação de estado para a região de vapor. Com esta equação, pode-se obter o
volume específico do vapor sobreaquecido, para dadas temperaturas e pressões.
- Determinar a entalpia e a entropia.
99
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Para calcular a entalpia de vaporização (hfg), utiliza-se a equação de Clapeyron (calcula-se o
declive da curva de saturação pela derivada da curva de saturação; usam-se os dados
experimentais do volume específico do líquido saturado; o valor do volume específico do vapor
saturado a uma dada temperatura obtém-se a partir da curva de saturação de vapor e da equação
de estado). A partir da entalpia de vaporização, obtém-se a variação de entropia:
s fg =
h fg
T
- Seguindo uma isotérmica na região de vapor sobreaquecido, a partir de um estado sobre a curva
de saturação do vapor, podemos determinar a entalpia e a entropia em outros pontos dessa
isotérmica utilizando as expressões vistas anteriormente:
p2⎡
⎛ ∂v ⎞ ⎤
h 2 − h 1 = ∫ ⎢ v − T⎜ ⎟ ⎥ dp
⎝ ∂T ⎠ p ⎦⎥
⎢
p1⎣
p2
⎛ ∂v ⎞
s 2 − s1 = − ⎜ ⎟ dp
⎝ ∂T ⎠ p
p1
∫
- Se seguirmos uma isobárica na região de vapor sobreaquecido, temos de usar as expressões
seguintes:
T2
h 2 − h1 = ∫ c p dT
T1
T2
s 2 − s1 =
∫ cp
T1
dT
T
11.4.3. Utilização das tabelas para líquido saturado e vapor saturado
As tabelas A4 e A5 (de Y. A. Çengel & M. A. Boles, 2006) do Anexo II fornecem, para
diferentes temperaturas (pressões), os valores da pressão de saturação (temperatura de saturação),
o volume específico, a energia interna específica, a entalpia específica e a entropia específica.
Vejamos alguns exemplos de aplicação dessas tabelas:
(i) Cálculo do calor latente de vaporização
O calor latente de vaporização de uma substância corresponde à quantidade de calor que a
unidade de massa dessa substância absorve durante a transição de fase líquido-vapor. Como a
transição de fase se dá a pressão constante, a variação da entalpia específica corresponde a essa
quantidade de calor:
dh = T ds + v dp = T ds (num processo isobárico)
Na tabela vemos que para a temperatura de 90ºC, a pressão de saturação é 70,183 kPa. Se a
vaporização se der nessas condições, o calor latente pode ser calculado do seguinte modo:
h(vapor sat.) - h(líq. sat.) = 2659,6 - 377,04 = 2282,6 kJ kg-1
(ii) Um tanque rígido contém 50 kg de água líquida saturada a 90ºC. Determine a pressão no
tanque e o seu volume.
Da tabela, psat = 70, 183 kPa e o volume específico do líquido saturado vf = 0,001036 m3 kg-1
100
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Então o volume total é: V = mv = (50) (0,001036) = 0,0518 m3
(iii) Uma massa de 200 g de água líquida saturada é completamente vaporizada à pressão
constante de 100 kPa. Determine a variação de volume e a quantidade de energia que foi
transferida para a água.
Da tabela, obtemos os valores do volume específico do líquido saturado e do vapor saturado e
subtraímos:
vg – vf = 1,6941 – 0,001043 = 1,6931 m3 kg-1
Então, a variação do volume será: (0,2 kg) (1,6931 m3 kg-1) = 0,3386 m3
A energia necessária para se dar a vaporização da unidade de massa é a entalpia de vaporização,
que àquela pressão (100 kPa) é 2257,5 kJ kg-1.
Então a energia total necessária será: ΔH = (0,2 kg) (2257,5 kJ kg-1) =451,5 kJ
Durante o processo de vaporização, uma substância existe parcialmente no estado líquido e
parcialmente no estado de vapor. A razão entre a massa de vapor (mg) e a massa total da mistura
(massa de vapor + massa de líquido) é definida como a qualidade x:
m vapor
mg
x=
=
m total
mf + mg
Considere-se que as duas fases estão bem misturadas formando uma mistura homogénea. Então
as propriedades desta mistura são as propriedades médias da mistura do líquido com vapor
saturado. Se um recipiente contiver uma mistura de líquido saturado e vapor saturado, o volume
total é a soma dos volumes de cada fase saturada. Mas o volume total é dado pelo produto da
massa total pelo volume específico médio da mistura. Então:
mt vméd = mf vf + mg vg = (mt - mg) vf + mg vg
Dividindo por mt:
vméd = (1 – x) vf + x vg = vf + x (vg - vf) = vf + x vfg
v
− vf
x = méd
v fg
p ou T
vméd = vf + x vfg
Um resultado semelhante pode ser obtido
também para a energia interna ou para a
entalpia:
uméd = uf + x ufg
vf vméd
hméd = hf + x hfg
vg
v
(iv) Um tanque rígido contém 10 kg de água a 90ºC. Se 8 kg estiverem na fase líquida e o resto
na fase de vapor, determine a pressão no tanque e o volume do tanque. Determine a qualidade x
da mistura e o volume específico médio.
A pressão é a pressão de saturação para aquela temperatura p = psat (90ºC) = 70,183 kPa
O volume total é a soma dos volumes parciais:
101
TERMODINÂMICA APLICADA
2009 Isabel Ambar
V = (8 kg) (0,001036 m3 kg-1) + (2 kg) (2,3593 m3 kg-1) = 4,73 m3
2
x=
= 0,2
10
vméd = vf + x vfg = 0,001036 + (0,2) (2,3593-0,001036) ≈ 0,4727 m3 kg-1
(v) Um recipiente de 80 L contém 4 kg de refrigerante-134a à pressão de 160 kPa. Determine a
temperatura, a qualidade e a entalpia do refrigerante e o volume ocupado pela fase gasosa.
Como não sabemos o estado (líquido, gasoso ou mistura líquido-gasoso) do refrigerante, vamos
comparar uma propriedade que esteja nos dados com os valores da fase líquida saturada e fase
gasosa saturada para a pressão dada.
v = V/m = 0,080 m3/4 kg = 0,02 m3 kg-1
A partir da tabela A11 (ver Anexo II) para o refrigerante-134a saturado à pressão 160 kPa, temos:
vf = 0,0007437 m3 kg-1
vg = 0,12359 m3 kg-1
Como vf < v < vg, o refrigerante está na região de mistura saturada. Então a temperatura é a de
saturação para aquela pressão: T = Tsat (160 kPa) = -15,60 ºC
A qualidade x é:
x=
v − vf
0,02 − 0,0007437
=
= 0,158
v fg
0,12359 − 0,0007437
h = hf + x hfg = 31,21 + (0,158) (209,90 kJ kg-1) = 64,4 kJ kg-1
Para calcular o volume ocupado pelo vapor podemos partir do valor do volume específico e
multiplicar pela massa de vapor. Para calcular a massa de vapor, vamos usar a própria definição
de qualidade:
mg = x mt = (0,158) (4 kg) = 0,632 kg
Então o volume de vapor é: Vg = mg vg = (0,632 kg) (0,12359 m3 kg-1) = 0,0781 m3 (ou seja, 78,1
L)
O volume de líquido é: 80 – 78,1 = 1,9 L
11.4.4. Utilização das tabelas para vapor sobreaquecido
Vamos dar alguns exemplos de aplicação das tabelas de vapor sobreaquecido.
(i) Determinar a energia interna da água a 200 kPa e 300ºC.
À pressão de 200 kPa, a temperatura de saturação é 120,21ºC. Como a temperatura a que está a
água excede a temperatura de saturação, a água está num estado de vapor sobreaquecido. Pelas
tabelas correspondentes (ver Anexo III) obter-se-ia um valor de u = 2808,8 kJ kg-1.
(ii) Determinar a temperatura da água num estado em que p = 0,5 MPa e h = 2890 kJ kg-1.
Pelas tabelas (Anexo III) vemos que para a pressão de 0,5 MPa, a entalpia de saturação é 2748,1
kJ kg-1. Como a entalpia da água é maior do que este valor, significa que está no estado de vapor
sobreaquecido. Fazendo a interpolação a partir dos valores da tabela, a temperatura obtida é
216,3ºC:
T (ºC)
200
250
H (kJ kg-1)
2855,8
2961,0
102
TERMODINÂMICA APLICADA
2009 Isabel Ambar
11.5. Ciclos de vapor
11.5.1. Ciclo de vapor de Carnot
Como vimos anteriormente, o rendimento do ciclo de gás de Carnot é o maior de entre todos os
ciclos trabalhando entre as mesmas duas temperaturas. Vamos considerar agora o desempenho de
um ciclo de vapor ideal (de Carnot) a operar dentro da região de saturação de uma substância
simples.
T
O fluido é aquecido reversivelmente a
temperatura constante numa caldeira (processo
1→2 na figura), é expandido isentropicamente
1 caldeira 2
numa turbina (processo 2→3), condensado
turbina
compressor
reversivel e isotermicamente num condensador
4
3
condensador
(processo
3→4)
e
comprimido
isentropicamente por um compressor (processo
s
4→1).
Há diversos inconvenientes associados a este ciclo:
(i) A limitação do ciclo à região de saturação obriga a que a temperatura máxima que pode ser
usada no ciclo tem de ser inferior à temperatura crítica (que, na água, é 374ºC). Mas limitar a
temperatura máxima implica limitar o rendimento do ciclo.
(ii) No processo de expansão isentrópica (2→3), a qualidade da mistura saturada diminui, i.e., a
turbina tem de trabalhar com quantidades crescentes de gotas de líquido misturada com o
vapor e isso prejudica o seu funcionamento (as gotas provocam erosão das lâminas). O
problema pode ser eliminado utilizando um fluido com uma curva de vapor saturado muito
inclinada.
(iii) A compressão isentrópica (4→1), envolve a compressão de uma mistura líquido-vapor até
ficar líquido saturado. Isto não é fácil de controlar e, além disso, não é prático um
compressor que trabalhe com as duas fases.
2
1
T
Alguns destes problemas podiam ser eliminados
realizando o ciclo de Carnot de modo diferente, como
4
3
ilustrado na figura junta. Contudo, isto traria outras
dificuldades (compressão isentrópica a altas pressões
e aquecimento isotérmico a pressões variadas).
s
11.5.2. Ciclo de vapor de Rankine
Muitas das dificuldades encontradas com o ciclo de Carnot podem ser eliminadas sobreaquecendo
o vapor na caldeira e condensando completamente o vapor no condensador.
O ciclo obtido é o ciclo de Rankine, que é o ciclo ideal para uma central térmica a vapor. O ciclo
ideal de Rankine não envolve qualquer processo internamente irreversível e consiste nos seguintes
quatro processos:
103
TERMODINÂMICA APLICADA
2009 Isabel Ambar
T
1→2: Compressão isentrópica numa bomba
3
2→3: Aquecimento isobárico numa caldeira
2
3→4: Expansão isentrópica numa turbina
1
4
4→1: Arrefecimento isobárico num condensador.
A água entra como líquido saturado na bomba no estado 1 e é comprimida isentropicamente até à
pressão a que opera a caldeira. A temperatura da água aumenta um pouco neste processo.
No estado 2, a água entra na caldeira como um líquido comprimido e sai como um vapor
s
sobreaquecido (estado 3).
O vapor sobreaquecido entra na turbina onde é expandido isentropicamente produzindo trabalho
(faz rodar o eixo de um gerador eléctrico). A pressão e temperatura do vapor baixam durante este
processo até ao estado 4.
No estado 4, a mistura líquido-vapor saturados (com alta qualidade) entra no condensador e vai
ser condensada isobaricamente perdendo calor para um meio refrigerante (como um lago, um rio
ou a atmosfera) até chegar ao estado 1 de líquido saturado.
Como vimos anteriormente, a área limitada por uma curva num diagrama T-s representa a
transferência de calor para processos internamente reversíveis. Então a área sob a curva 2-3
representa o calor transferido para a água na caldeira e a área sob a curva 4-1 representa o calor
perdido pela água no condensador. A diferença entre estas duas quantidades, isto é, a área
envolvida pelo ciclo, corresponde ao trabalho resultante durante o ciclo (trabalho realizado pela
turbina menos o trabalho recebido da bomba).
O rendimento térmico de um ciclo de Rankine é dado pela razão seguinte:
Wres
η=
Q receb
em que Wres é o trabalho resultante (trabalho realizado pela turbina – trabalho recebido na bomba)
e Qreceb é o calor recebido na caldeira.
Como ΔU = Q + W, vem 0 = |Qreceb| - |Q perd| + |Wreceb| – |W realiz|
Wres = |W realiz| - |Wreceb| = |Qreceb| - |Q perd|
Então:
Q perd
Wres
η=
= 1−
Q receb
Q receb
Um ciclo de Carnot a trabalhar entre as mesmas temperaturas que o ciclo de Rankine teria um
rendimento muito maior.
104
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Exercício:
Considere uma central térmica a operar segundo um ciclo de Rankine ideal. O fluido entra na
turbina a 3 MPa e 350ºC e é condensado no condensador à pressão de 75 kPa. Calcular:
(i)
Entalpia do estado 1
Estado 1: Líquido saturado, pressão de 75 kPa.
Pelas tabelas,
h1 = 384,44 kJ kg-1
(ii) Entalpia do estado 3
Estado 3: Vapor sobreaquecido, pressão de 3 MPa e
temperatura 350ºC. Pelas tabelas:
h3 = 3115,1 kJ kg-1
(iii) Entalpia do estado 4
Estado 4: Mistura líquido-vapor saturados, qualidade x
h4 = hf + x4 hfg
Para calcular a qualidade x, temos de utilizar outra propriedade de que se conheçam os
valores: podemos usar a entropia visto que s4 = s3 e o valor de s3 pode ser obtido a partir das
tabelas de vapor sobreaquecido (para p=3 MPa e T=350ºC)
s3 = 6,7450 kJ kg-1 K-1
A qualidade x4 = (s4-sf)/sfg = (6,7450 – 1,2132)/6,2426 = 0,8861
Para p=75 kPa, hf = 384,44 kJ kg-1 e hfg=2278,0 kJ kg-1.
Então:
h4 = hf + x4 hfg = 384,44 + 0,8861 (2278,0) = 2403,0 kJ kg-1
Bibliografia
Physics for Scientists and Engineers. Serway, Saunders College Pub., (4ª edição) 1982.
Thermodynamics. An Engineering Approach. Y. A. Çengel & M. A. Boles, McGraw Hill (5ª
edição), 2006.
Thermodynamics and an Introduction to Thermostatics. H.B. Callen, J. Wiley, (2ª edição) 1985.
Fundamentos de Termodinâmica do Equilíbrio. J. Güemez, C. Fiolhais & M. Fiolhais, Fundação
C. Gulbenkian, 1998.
Thermodynamics. K. Wark, McGraw-Hill, (3ª edição), 1977.
Heat and Thermodynamics. M.W. Zemansky & R. H. Dittman, McGraw-Hill (6ª edição), 1981.
Fundamentals of Classical Thermodynamics. G. van Wylen, R. Sonntag & C. Borgnakke, J.
Wiley, (4ª edição), 1994.
Principles of General Thermodynamics. Hatsopoulos & Keenan, J. Wiley, 1965.
Anexos
Anexo I - Tabelas com o coeficiente de expansão térmica, compressibilidade isotérmica e
calor específico da água do mar em função da temperatura, salinidade e pressão.
Anexo II – Tabelas com propriedades termodinâmicas de líquido e vapor saturados
Anexo III - Tabelas com propriedades termodinâmicas de vapor de água sobreaquecido e
água líquida comprimida
105
TERMODINÂMICA APLICADA
2009 Isabel Ambar
ANEXO I
Tabelas com a variação dos coeficientes de expansão térmica, compressibilidade isotérmica
e calor específico da água do mar em função da temperatura, da salinidade e da pressão.
106
TERMODINÂMICA APLICADA
2009 Isabel Ambar
107
TERMODINÂMICA APLICADA
2009 Isabel Ambar
108
TERMODINÂMICA APLICADA
2009 Isabel Ambar
109
TERMODINÂMICA APLICADA
2009 Isabel Ambar
110
TERMODINÂMICA APLICADA
2009 Isabel Ambar
-
Tabelas com o valor do calor específico de vários gases em função da temperatura
(Thermodynamics. An Engineering Approach. Y. A. Çengel & M. A. Boles, McGraw
Hill, 2006).
111
TERMODINÂMICA APLICADA
2009 Isabel Ambar
112
TERMODINÂMICA APLICADA
2009 Isabel Ambar
ANEXO II
Tabelas com propriedades termodinâmicas de líquido e vapor saturados
113
TERMODINÂMICA APLICADA
2009 Isabel Ambar
114
TERMODINÂMICA APLICADA
2009 Isabel Ambar
115
TERMODINÂMICA APLICADA
2009 Isabel Ambar
Anexo III
Tabelas com propriedades termodinâmicas de vapor de água sobreaquecido e água líquida
comprimida
116
TERMODINÂMICA APLICADA
2009 Isabel Ambar
117
TERMODINÂMICA APLICADA
2009 Isabel Ambar
118