1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
SUMÁRIO
PAG.
HISTÓRIA DA GENÉTICA
01
DAS ERVILHAS A DUPLA HÉLICE
01
GÉNETICA
10
O ESTUDO DE MENDEL SOBRE A HEREDITARIEDADE
10
O ORGANISMO EXPERIMENTAL DE MENDEL, A ERVILHA
10
O PRINCÍPIO DA DOMINÂNCIA
15
O PRINCÍPIO DA SEGREGAÇÃO:
15
O PRINCÍPIO DA DISTRIBUIÇÃO INDEPENDENTE:
17
APLICAÇÕES DOS PRINCÍPIOS DE MENDEL
18
O MÉTODO DO QUADRADO DE PUNNETT
18
O MÉTODO DA LINHA BIFURCADA
18
O MÉTODO DA PROBABILIDADE
19
FORMULAÇÃO E TESTE DAS HIPÓTESES GENÉTICAS
21
O TESTE DO QUI-QUADRADO
22
PRINCÍPIOS MENDELIANOS EM GENÉTICA HUMANA
24
HEREDOGRAMAS
25
SEGREGAÇÃO MENDELIANA EM FAMÍLIAS HUMANAS
26
CONSULTA GENÉTICA
28
RELEVANCIA DOS GENES E CROMOSSOMOS
31
CROMOSSOMOS
31
NÚMERO DE CROMOSSOMOS
31
CROMOSSOMOS SEXUAIS
32
TEORIA CROMOSSÔMICA DA HERANÇA
33
CROMOSSOMOS COMO VEÍCULOS DE GENES
35
NÃO-DISJUNÇÃO COMO PROVA DA TEORIA CROMOSSÔMICA
37
A BASE CROMOSSÔMICA DOS PRINCÍPIOS MENDELIANOS DA
39
SEGREGAÇÃO E DA DISTRIBUIÇÃO INDEPENDENTE
O PRINCÍPIO DA SEGREGAÇÃO
39
O PRINCÍPIO DA DISTRIBUIÇÃO INDEPENDENTE
41
GENES LIGAODOS AO SEXO EM SERES HUMANOS
41
HEMOFILIA, UM DISTÚRBIO DE COAGULAÇÃO SANGUÍNEA LIGADO AO X
42
DALTONISMO, UM DISTÚRBIO VISUAL LIGADO AO X
42
A SÍNDROME DO X FRÁGIL E RETARDO MENTAL
44
GENES NO CROMOSSOMO Y HUMANO
45
GENES NOS CROMOSSOMOS X E Y
46
CROMOSSOMOS SEXUAIS E DETERMINAÇÃO DO SEXO
46
DETERMINAÇÃO DO SEXO EM SERES HUMANOS
46
DETERMINAÇÃO DO SEXO NA DROSOPHILA
48
DETERMINAÇÃO DO SEXO EM OUTROS ANIMAIS
49
COMPENSAÇÃO DE DOSE DE GENES LIGADOS AO X
51
HIPERATIVAÇÃO DE GENES LIGADOS AO X EM MACHOS DE DROSOPHILA
52
INATIVAÇÃO DE GENES LIGADOS AO X EM FÊMEAS DE MAMÍFEROS
52
GENÉTICA EM MEDICINA
55
GENÉTICA E AGRICULTURA MODERNA
59
GENÉTICA E SOCIEDADE
62
0
História da Genética
Das ervilhas a dupla hélice
Adaptado de Barros Veloso, 2003
1900 foi o ano que, separadamente, três biólogos – Hugo de Vries, Erich von Tschermak e
Karl Correns –descobriram os trabalhos de Mendel que se encontravam esquecidos há 35 anos nas
estantes de velhas bibliotecas. Este fato iria marcar a ciência e a tecnologia do século XX. A
redescoberta das leis mendelianas forneceu a base conceptual para o desenvolvimento da genética
que, ao culminar, 53 anos mais tarde, na dupla hélice de Watson e Crick, abriria caminho à
engenharia genética, à clonagem e à decifração do genoma.
Nos 10 anos anteriores, os avanços da microscopia tinham tornado possível observar os
cromossomas e os fenômenos de meiose. “Ora, as idéias desenvolvidas por Mendel sobre os fatores
hereditários” transmitidos pelas células reprodutoras e a sua combinação aleatória quando da
fecundação, estavam de acordo com os dados fornecidos pela fisiologia celular que a microscopia
começara a revelar. Foi precisamente esta semelhança entre as teorias de Mendel e o
comportamento dos cromossomas que permitiu a Walter Sutton, em 1902, propor a primeira teoria
cromossômica da hereditariedade. Mas tudo isto aconteceu porque os trabalhos de Mendel tinha
sido exemplo genial de concepção e de método.
Nascido em 1822, Mendel foi orientado para uma carreira eclesiástica a fim de poder
continuar os seus estudos e, depois de freqüentar em Viena o curso de biologia e de física durante
dois anos, foi nomeado superior do mosteiro agostiniano de Brno. Aí, num pequeno jardim de 35
por 7 metros, que ainda hoje pode ser visitado, realizou os seus trabalhos de hibridação com
ervilheiras.
Mendel percebeu também que seria importante realizar as suas observações numa população
numerosa de plantas, a fim de fazer um tratamento estatístico dos resultados. Desta maneira, estava
a aplicar pela primeira vez à biologia o rigor da matemática. Além disso, teve a intuição de recorrer
à utilização de símbolos (A, dominante, Aa, híbrido e a, recessivo) com os quais pôde articular a
teoria com a experimentação. Ele tinha percebido que uma coisa é o que se vê – o caráter –outra as
partículas ou unidades ocultas – os “fatores” – que se exprimem por sinais exteriores. Desta forma
estava a antecipar os conceitos de “fenótipo” e “genótipo” que a genética iria consagrar mais tarde.
Mas a verdade é que, mesmo quando o desenvolvimento da genética veio mostrar que a
transmissão de caracteres era mais complexa do que Mendel alguma vez terá pensado, as suas leis
se revelaram rigorosamente exatas. Bastou para isso acrescentar algumas “extensões ao
1
mendelismo”, tais como a “dominância intermédia” (flores cor-de-rosa provenientes de
progenitores com flores vermelhas e flores brancas), a “co-dominância” (grupos sanguíneos), a
“interação gênica”, etc.
De Vries, trabalhando com uma planta da família das Onagraceae, deu o passo seguinte, ao
descobrir modificações bruscas, descontinuas e hereditárias, capazes de alterar caracteres. Chamoulhes “mutações”, mas sabe-se hoje que o que ele observou foram, sobretudo, acidentes
cromossômicos heterogêneos e pouco freqüentes que não eram reprodutíveis em outras variedades
de plantas. Tal como Mendel, De Vries generalizou aquilo que era exceção. Mas, ao fazê-lo,
introduziu um novo conceito que se revelaria fundamental no desenvolvimento posterior da
genética.
Em 1910, Morgan observou pela primeira vez a presença de um macho de Drosophila
melanogaster que tinha olhos brancos, em vez dos habituais olhos vermelhos. Aplicando então os
métodos que tinham sido utilizados por Mendel, verificou que, na primeira geração resultante do
cruzamento entre esse macho e uma fêmea normal,( todas com olhos vermelhos). Contudo, na
segunda geração, todas as fêmeas apresentavam olhos vermelhos enquanto que, em metade dos
machos, os olhos eram brancos. Foi o estudo destas populações que lhe permitiu concluir a
existência de um caráter recessivo situado no cromossomo sexual. Estava assim encontrada a
primeira localização cromossômica para um fator hereditário mendeliano. Morgan em breve
detectou outras mutações.
A transmissão não independente de algumas delas permitiu-lhe admitir a existência de
linkage de vários genes que se exprimiam em conjunto, ao constatar o aparecimento no mesmo
macho de dois caracteres recessivos resultantes de mutações (olhos brancos e asas rudimentares),
localizados no cromossomo X, admitiu a hipótese de crossing-over, ou seja, de troca de material
genético entre os cromossomos do mesmo par. O colaborador, Alfred Sturtevant, sugeriram que,
quanto mais afastados estivessem os genes no mesmo cromossomo, maior seria a probabilidade de
se separarem numa geração posterior pelo mecanismo de crossing-over. A partir desta observação
foi possível desenvolver um vasto trabalho que permitiu definir a distribuição linear de alguns genes
ao longo dos quatro pares de cromossomos da drosófila. Surgia assim o esboço da primeira carta
genética, em que cada gene correspondia a um locus. Ao fim de duas décadas, foi referenciados
mais de 2500 genes. A teoria cromossômica da hereditariedade de Morgan é um marco na história
da genética, realizou uma síntese perfeita entre genética mendeliana e biologia celular. Sabiam que
na constituição dos cromossomos entravam proteínas e ácidos nucléicos.
2
O percurso que levou a atribuir ao ADN um papel central na transmissão dos caracteres
hereditários teve início e não foi simples. Recorde-se que a história do ADN tinha começado em
1868 quando um jovem bioquímico suíço, Johannes Miescher, conseguiu, pela primeira vez, separar
o núcleo do citoplasma. Isolou então, no núcleo dos espermatozóides do salmão, uma substância a
que chamou nucleína, que, além de proteínas, continha um composto rico em fósforo. Quando se
identificaram todos os constituintes químicos deste composto – fosfato, açúcar, bases púricas e
pirimídicas – passaram-se a chamar-lhe ácido desoxirribonucléico. Mas nada se conhecia acerca das
funções que desempenhava e ninguém parecia disposto a atribuir-lhe qualquer papel de destaque na
transmissão genética.
Nas primeiras décadas do século XX existia como que uma espécie de “imperialismo
conceptual” favorável às proteínas que só foi ultrapassado por uma série de trabalhos experimentais
que ficaram célebres pelo seu rigor e elegância. Vejamos, então, o que se passou. Em 1928, um
médico inglês, Fred Griffith, injetou num rato pneumococos não patogênicos juntamente com uma
suspensão de pneumococos patogênicos inativados pelo calor. Ao contrário do que seria de esperar
o rato morreu em 24 horas, tendo o exame do sangue revelado uma proliferação de bactérias
virulentas.
A questão que se levantou foi a de saber qual seria a substância, presente na suspensão de
bactérias inativadas, capaz de induzir este comportamento inesperado nas estirpes não patogênicas.
Griffith, que não tentou investigar se a agressividade bacteriana se perpetuava ou não nas gerações
seguintes, atribuiu este fenômeno a uma substância nutritiva.
Só em 1935 é que um médico canadiano, Oswald Avery, que trabalhava no Instituto
Rockfeller, decidiu resolver o enigma deixado em suspenso por Griffith. Com a ajuda de dois
colaboradores (Colin MacLeod e Maclyn McCarthy) procurou identificar o “fator transformante”,
responsável pela mudança operada nos pneumococos não patogênicos. Foi um longo e minucioso
trabalho realizado ao longo de 10 anos, com grande rigor e persistência, em que todas as técnicas
utilizadas conduziram ao mesmo resultado: o “fator transformante” não era uma proteína, mas sim o
ácido desoxirribonucléico. De fato, nos ensaios realizados, ficou demonstrado que o “fator
transformante” resistia a temperaturas que desnaturavam as proteínas; que após purificação por
testes colorimétricos, o “fator transformante” não continha ARN, nem proteínas, mas apena ADN;
que não era destruído nem pelas proteases que fragmentavam as proteínas, nem pelas fosfatases que
degradavam o ARN; e que era inativado pelo soro não aquecido, o qual possuía enzimas capazes de
3
destruir o ADN. Além disso, as análises químicas elementares mostravam que o “fator
transformante” purificado continha quantidades de proteínas muito inferiores a 1%.
Avery dedicou ainda uma parte importante do seu trabalho ao estudo imunológico do
material isolado, verificando que ele não reagia com os anticorpos anticápsula bacteriana. Tudo
levava a crer que possuía uma composição química diferente da estrutura celular que era suposto
transformar. Contudo, o que mais contribuiu para a falta de aceitação destes resultados, foi a
convicção, então generalizada entre os geneticistas, de que só as proteínas podiam veicular a
transmissão de caracteres hereditários.
Os dados apresentados não se inseriam nos conhecimentos nem nos consensos da época,
pelo que ninguém sabia “o que fazer das observações de Avery”, como teria dito na altura Max
Delbrück. De facto não era fácil admitir que um ácido nucleico, composto que não revelava
qualquer especificidade quando submetido aos critérios imunoquímicos, pudesse controlar a
atividade de proteínas ou, mais precisamente, de enzimas responsáveis pela síntese da cápsula de
pneumococos. Mas outro acontecimento viria contribuir para desvalorizar os resultados de Avery e
para reforçar a posição dos “proteinófilos”. No princípio da década de 40, Beadle e Tatum iniciaram
um trabalho com um fungo, a Neurospora, cujo ciclo evolutivo permitia um rápido isolamento de
mutantes.
Estes trabalhos permitiram pela primeira vez estabelecer uma associação entre bioquímica e
genética e impuseram a relação simplista “um gene-um enzima”. E, naturalmente, reforçaram os
argumentos a favor do papel desempenhado pelas proteínas na hereditariedade. Mas, enquanto tudo
isto se passava, outros acontecimentos preparavam novos rumos para a genética através da
aproximação da fisica à biologia.
A atividade dos físicos passou a resumir-se à verificação e ao aperfeiçoamento de modelos
já consagrados, sem que fossem postos em causa os fundamentos conceptuais da disciplina. Neste
contexto, a biologia surgiu como uma “nova fronteira” do conhecimento à qual a física quântica
poderia fornecer novos e valiosos utensílios e onde jovens físicos poderiam encontrar uma área
estimulante para a sua atividade. Um dos físicos que mais influenciou a biologia foi Max Delbrück.
Nascido em 1906, em Berlim, defendeu tese de doutoramento em física teórica em 1930. Em 1932,
durante um estágio em Copenhaga, assistiu a uma conferência de Niels Bohr (“Light and Life”) que
muito o impressionou. Bohr era o papa da mecânica quântica, o chefe de fila da “interpretação de
Copenhaga” e o autor do polémico “princípio da complementaridade”. A mensagem retida por
Delbrück terá sido esta: é necessário levar o estudo molecular dos seres vivos tão longe quanto
4
possível e há que fazer uma abordagem diferente da vida, que seja “complementar” daquela que
estava a ser feita até então.
Tal como os princípios da mecânica quântica só tinham sido revelados quando a matéria
passou a ser estudada ao nível mais elementar – o átomo –, também para descobrir os segredos da
vida era necessário desvendar os sistemas biológicos mais simples. De regresso a Berlim, Delbrück
empenhou-se em aplicar à biologia os modelos do bombardeamento do átomo utilizados pela física,
expondo a drosófila aos efeitos dos raios X. O seu objetivo era relacionar o número de mutações
com a energia das radiações utilizadas. Pôde, assim, estabelecer um modelo quântico do gene que,
tal como uma molécula, possuiria vários níveis estáveis de energia: uma mutação não seria mais do
que a passagem de um estado estável a outro estado estável. Da mesma forma que as variações da
matéria e da energia, também as variações hereditárias se fariam por “saltos quânticos”. O artigo
datado de 1935, em que publicitou estas experiências, chegou às mãos de Erwin Schrödinger, um
dos físicos que mais contribuiram para o desenvolvimento da mecânica quântica.
Foi ele que num livro, “What is Life?”, deu ampla publicidade aos trabalhos de Delbrück e
admitiu que os genes têm de ser necessariamente constituídos por um número limitado de átomos,
cujo ordenamento é capaz de reproduzir uma variedade infinita de configurações espaciais e
funcionais. Sugeriu pela primeira vez a existência de um código genético, hipótese que, embora
especulativa, viria a constituir o quadro teórico de toda a investigação posterior. Ao contrário de
Bohr, que adotara uma interpretação indeterminista, Schrödinger bateu-se por uma ordem e uma
lógica determinista dos seres vivos, cujas leis próprias ele admitiu que estavam ainda por esclarecer.
Quando Delbrück passou pelo laboratório de Thomas Morgan, em Pasadena, no ano de
1937, estava já convencido que a drosófila era um sistema complexo demais para o estudo dos
segredos da vida. Seria necessário, por isso, procurar sistemas elementares mais simples. Foi assim
que escolheu os bacteriógafos, descobertos em 1917 por Félix d’Herelle, que lhe pareciam ser
partículas biológicas elementares e que estariam, por isso, para os sistemas biológicos, como as
moléculas para a matéria. Entretanto um italiano, Salvador Luria, que trabalhara com Enrico Fermi
em Roma, viu-se forçado a fugir para os EUA em 1941, tendo começado a colaborar com Delbrück
em Cold Spring Harbor. Quando, em 1943, a eles os dois se juntou Alfred Hershey formou-se o
chamado “grupo do fago” que iria procurar esclarecer os mistérios ligados à replicação dos
bacteriófagos. A importância deste grupo ficou a dever-se, em grande parte à personalidade
carismática de Delbrück, aos seus métodos revolucionários de trabalho e à sua forte convicção de
5
que os mecanismos de reprodução dos seres vivos eram os mesmos, quer se tratasse de vírus ou de
animais superiores.
Em breve o trabalho do grupo iria traduzir-se em realizações concretas. Luria, ao estudar a
resistência das bactérias a um fago, verificou que não eram os fagos que induziam resistências:
apenas selecionavam as bactérias previamente resistentes. O tratamento matemático destes dados,
realizado por Delbrück, permitiu, pela primeira vez, calcular taxas de mutação, que constituiram
uma informação essencial para uma análise genética. Mas esta experiência demonstrou, sobretudo,
a origem mutacional das estirpes resistentes e a importância da seleção na evolução dos seres vivos.
Constituiu, assim, uma importante vitória do darwinismo que, a partir daí, ficou definitivamente
ligado à biologia molecular. Mas a realização mais importante do “grupo do fago” foi conseguida
em 1952 por Alfred Hershey e Martha Chase numa série de experiências em que, pela primeira vez
em biologia, foi utilizada a marcação de moléculas com substâncias radioactivas. Em trabalhos
realizados anteriormente tinha ficado claro que o bacteriófago era constituído por um invólucro
proteico que continha ADN no seu interior. Depois de marcados alternadamente com 35S (que se
incorpora nas moléculas proteicas) ou com 32P (que seintegra no ADN), os bacteriógados eram
depois adicionados a culturas bacterianas. Todos os resultados demonstraram que o ADN marcado
com 32P era introduzido, digamos
mesmo, injectado para dentro das bactérias, onde se replicava e dava origem a novos bacteriófagos.
Por sua vez, as proteínas marcadas com 35S permaneciam fora das bactérias.
Ficava assim demonstrado que o ADN era o material genético e que as proteínas apenas
desempenhavam, neste caso, um papel de invólucro estrutural. Estes resultados foram então
apresentados como a primeira prova do papel genético desempenhado pelo ADN. Mas a verdade é
que oito anos antes, Avery, utilizando métodos diferentes, tinha chegado à mesma conclusão. E se,
na altura, Avery fora criticado pela interpretação pouco rigorosa que tinha feito de alguns
resultados, muito mais razões haveria para questionar a falta de rigor de Hershey e Chase. O
problema é que, durante esses oito anos, outros dados se tinham acumulado e, a pouco e pouco, os
resultados apresentados por Avery começavam a pesar no espírito dos investigadores. Foi assim
que, entre 1944 e 1952, a comunidade científica foi sendo preparada para aquilo a que Thomas
Kuhn, na “Estrutura das Revoluções Científicas”, iria chamar mais tarde uma “conversão”. A
disparidade com que os geneticistas trataram Avery, por um lado, e Hershey e Chase, por outro,
apenas demonstra que uma Linus Pauling – Prémio Nobel da Química em 1954, Prémio Nobel da
Paz em 1962, olhado com suspeição pelo mccarthysmo – foi uma das figuras mais brilhantes do
6
século XX. Foi ele que, adaptando os conceitos da mecânica quântica ao estudo das moléculas,
mostrou que era possível prever as ligações químicas a partir da estrutura eletrônica dos átomos.
Juntando a isto os dados obtidos pelos estudos cristalográficos, estabeleceu um conjunto de regras
simples que permitiam “adivinhar” a estrutura espacial das moléculas. Por isso a ele se ficaram a
dever aqueles sugestivos objetos constituídos por agregados de esferas de várias cores que povoam
as páginas dos modernos tratados de biologia.
Mas Pauling também percebeu a importância das ligações fracas, ou ligações hidrogénicas,
na formação das estruturas biológicas. Funcionando como “botões de mola”, estas ligações iónicas
permitem uma adesão precisa, firme e ao mesmo tempo lábil das moléculas, desempenhando assim
um papel estabilizador nas estruturas tridimendionais das proteínas. Através delas tornou-se
possível compreender as interacções entre as macromoléculas, nas quais assenta a estrutura e o
funcionamento dos seres vivos. Foi assim que, em 1951, Pauling descreveu, pela primeira vez, a
estrutura helicoidal de certas cadeias polipeptídicas – nomeadamente a “hélice alfa” – que daí para
frente passaria a estar sempre presente na imaginação dos biólogos moleculares.
Na Inglaterra, a pesquisa sobre o ADN era, segundo Watson, “propriedade privada” do
físico Maurice Wilkins do King’s College, que utilizava a difração pelos raios X. Esta técnica,
permitia estudar a configuração tridimensional das macromoléculas biológicas, através de imagens
fotográficas obtidas quando um cristal da substância era exposto a um feixe de raios X. Watson e
Crick estavam, naturalmente, a par do que se passava, conheciam, além disso, os trabalhos que
Chargaff realizara na Universidade de Columbia, embora, na altura, não se percebesse qual poderia
ser a sua utilidade. Chargaff tinha analisado diversas amostras de ADN, tendo verificado que as
quantidades das bases aminadas variavam com as espécies, mas que, no mesmo organismo, a
quantidade de adenina era sempre igual à de timina e a quantidade de guanina era sempre igual à de
citosina.
Este dado iria ter, mais tarde, uma influência decisiva no esclarecimento da estrutura do
ADN. Não deixa de ser bizarro que Watson e Crick nunca tenham feito qualquer experiência que
envolvesse a molécula de ADN. O seu método de trabalho assentava, como já foi dito, em longas
discussões teóricas, durante as quais tentavam ultrapassar problemas e esclarecer as questões mais
obscuras. Ao mesmo tempo procuravam construir, com peças metálicas talhadas á medida,
estruturas tridimensionais que estivessem de acordo com os dados conhecidos. Tal como fizera
Pauling, tentavam entender de que maneira é que os átomos tinham tendência para se ligar uns aos
outros pois, como diria Watson, o que se impunha era construir “uma série de modelos moleculares
7
e começar a brincar com eles” como se fossem “brinquedos das crianças da pré-primária”. Mas
sempre na convicção, não partilhada por outros, de que a estrutura do ADN era uma hélice e que,
antes de pensar em modelos complexos, era necessário pôr à prova as soluções mais simples.
Como é sabido, Watson e Crick acabariam por encontrar a solução correta. Mas pelo meio
ficaria um incidente marcado pela falta de “fair play” que os tornaria alvo de muitas críticas. Tudo
aconteceu quando Wilkins, talvez ferido com a agressividade de Rosy do laboratório Inglês, revelou
a Watson dados recentes obtidos nolaboratório King’s College e que eram favoráveis a uma nova
forma tridimensional do ADN. Tratava-se de uma imagem de difracção a que tinham chamado
“estrutura B” que, além de ser “inacreditavelmente” mais simples do que a anterior (estrutura A), só
poderia corresponder a uma estrutura helicoidal. Além disso, a observação desta nova imagem
obtida com os raios X permitia, através de cálculos relativamente rápidos, obterem alguns dados
essenciais acerca da molécula. Mas Wilkins forneceu ainda mais uma informação: Rosy estava
agora convencida de que as bases aminadas se encontravam no centro da estrutura molecular,
envolvidas por um esqueleto exterior “açúcar-fosfato”. Watson ficou excitadíssimo com estas
revelações e começou imediatamente a trabalhar no novo modelo.
Havia que encomendar a um mecânico peças metálicas das purinas e das pirimidinas, assim
como dos átomos de fósforo, e aguardar pela sua montagem. Nessa altura não era ainda possível
saber se o ADN tinha duas se três cadeias, mas Watson, apesar das reservas de Crick, decidiu
começar a “jogar” com modelos de duas cadeias. Passados poucos dias tinham já construído uma
configuração estereoquímica para a molécula que estava de acordo com aquilo que tinham
imaginado. Mas o que então ninguém sabia é que também já dispunham de dados pormenorizados
acerca de toda a investigação que estava a ser feita por Rosy. A utilização abusiva desta informação,
que não tinha sido ainda divulgada e a que tiveram acesso de uma forma confidencial, constituiu
uma marca eticamente negativa do trabalho de Watson e Crick.
Um problema continuava, contudo, por resolver: o emparelhamento das bases. Pensava-se
nessa altura que as bases idênticas emparelhavam entre si (adenina com adenina, timina com timina,
etc.) e se uniam por ligações hidrogênio. Mas sendo assim, e uma vez que as purinas e as
pirimidinas tinham formas tautoméricas diferentes, o esqueleto da estrutura helicoidal ficaria
deformado para dentro ou para fora de acordo com os pares de bases que, em cada passo da
hélice, estivessem no centro. Foi Jerry Donohue – cristalógrafo americano que trabalhara com Linus
Pauling – que, ao notar que as formas tautoméricas com que Watson estava a trabalhar eram
incorretas, lhe chamou a atenção para um aspecto que se revelaria fundamental: o par guanina-
8
citosina tinha uma forma espacial idêntica ao par adenina-timina. Por isso o emparelhamento teria
sempre de ser feito entre estes pares de bases e não entre outros.
As peças do “puzzle” começavam a encaixar umas nas outras e pemitiam compreender os
dados obtidos por Chargaff que, na altura em que tinham sido divulgados, pareciam não fazer
qualquer sentido. Agora se tornava claro porque é que as quantidades de adenina e de timina tinham
de ser iguais, da mesma forma que iguais tinham de ser as quantidades de guanina e de citosina.
Watson e Crick estavam assim à beira de esclarecer um dos problemas mais importantes da
biologia. Sem uma única experiência laboratorial, recorrendo apenas a modelos conceptuais, tinham
encontrado o modelo de uma dupla hélice que era forçoso que fosse assim porque, como diria
Watson, “uma estrutura tão bonita tinha pura e simplesmente de existir”. A 25 de Abril de 1953 a
Nature publicava um artigo, com pouco mais de uma página, intitulado “Molecular structure of
nucleic acids”, no qual uma frase premonitória anunciava todo um programa posterior de
investigação genética: “It has not escaped our notice that the specific pairing we have postulated
immediately suggest a possible copying mechanism for the genetic material”.
Nesse mesmo ano o mundo assistia à coroação da Rainha Isabel II e à conquista do Everest.
Mas só um jornal britânico – o News Chronicle – se referia à dupla hélice num artigo intitulado
“Nearer secret of life”. Watson, Crick e Wilkins receberiam o Prémio Nobel em 1962. Rosalind
Franklin, cuja contribuição fôra fundamental para este feliz desfecho, não estava presente: tinha
falecido em 1958, aos 37 anos, com um cancro do ovário. Passados 50 anos, algumas consequências
desta descoberta são agora bem visíveis. A ciência, ao mesmo tempo que tem tentado desvendar os
mistérios da reprodução e da hereditariedade, fez também reacender velhos receios e temores.
E o Homem parece à beira de se apoderar, mais uma vez, de atributos que eram pertença exclusiva
das divindades: depois de dominar o fogo e de aprender a voar, prepara- se agora para controlar a
própria origem da vida. Ora, sempre que coisas destas acontecem, renascem os mitos cuja presença
é uma constante no nosso inconsciente coletivo. Quer sob a forma de andróginos-cortados-ao-meio
por Zeus, de Prometeu agrilhoado, de expulsão do Paraíso ou de caos poliglótico de Babel,
ressurgem de novo as imagens simbólicas que dão conteúdo e sentido ético à vida e à ação dos
homens. Mas, mais do que recear o castigo dos deuses, há que ter presente o risco de provocar
rupturas nos equilíbrios que estão subjacentes à própria natureza das coisas. E, em vez de condenar
a ciência e a tecnologia, alimentando temores irracionais, é altura de recuperar e reciclar os velhos
mitos, dando-lhes o significado que hoje têm, como equivalentes arcaicos que são, de um debate
ético que é preciso desenvolver e aprofundar
9
GÉNETICA
Adaptado de Snustad & Simmons, 2000.
O ESTUDO DE MENDEL SOBRE A HEREDITARIEDADE
A vida de Gregor Johann Mendel (1822-1884) cobriu metade do século XIX. Seus pais
eram fazendeiros na Moravia, então uma parte do império Habsburg na Europa Central. A vida
rural lhe ensinou a cuidar de plantas e animais e lhe inspirou o interesse pela natureza. Aos 21
anos, Mendel deixou a fazenda e entrou para um monastério católico na cidade de Brünn (hoje,
Brno, na república Tcheca). Em 1847, ele foi ordenado padre, adotando o nome de Gregor.
Subsequentemente ele lecionou na escola local, fazendo um intervalo entre 1851 e 1853 para
estudar na Universidade de Viena. Após retornar a Brünn, retomou sua vida de monge professor e
começou seus experimentos genéticos que por fim o tornaram famoso.
Mendel fez experimentos com várias espécies de plantas de jardim, e até tentou alguns
experimentos com abelhas. Seu maior sucesso, entretanto, foi com ervilhas. Completou seus
experimentos com ervilhas em 1863, e passou os dois anos seguintes analisando e resumindo seus
dados. Em 1865, Mendel apresentou os resultados à sociedade de história natural local, e no
ano seguinte publicou um relato detalhado nos anais da sociedade. Infelizmente, esta publicação
ficou na obscuridade até 1900, quando foi redescoberta por três botânicos — Hugo de Vries, na
Holanda; Cari Correns, na Alemanha, e Eric von Tschermak-Seysenegg, na Áustria. Quando eles
revisaram a literatura científica em busca de dados de apoio para suas próprias teorias sobre
hereditariedade, cada um deles descobriu que Mendel havia feito uma análise detalhada e cuidadosa
há 35 anos. As idéias de Mendel rapidamente ganharam aceitação, especialmente pelos esforços
promocionais de um biólogo britânico, Willian Bateson. Este campeão das descobertas de Mendel
criou um novo termo para descrever o estudo da hereditariedade: genética, da palavra grega que
sinifica “gerar”.
O Organismo Experimental de Mendel, a Ervilha
Um motivo do sucesso de Mendel foi a escolha astuta de seu material experimental. A
ervilha de jardim, Pisum sativum, é uma dicotiledônea, um tipo de planta que gera duas folhas, ou
cotilédones, a partir da germinação da semente. As ervilhas crescem facilmente em canteiros
experimentais ou em vasos em uma estufa.
10
Uma peculiaridade da reprodução de ervilhas é que as pétalas da flor se fecham firmemente,
impedindo que os grãos de pólen entrem ou saiam. Isto força um sistema de autofertilização, no
qual espermatozóides e ovócitos de uma determinada flor se unem para produzir as sementes. Como
resultado, linhagens individuais de ervilhas são altamente endogâmicas, apresentando pouca ou
nenhuma variação genética de uma geração para a seguinte. Devido a esta uniformidade,
dizemos que tais linhagens são true-breeding.
No final, Mendel obteve muitas variedades puras de ervilhas, cada uma se distinguindo por
uma característica particular. Em uma linhagem, as plantas tinham entre 180 cm e 212 cm,
enquanto em outras mediam apenas de 228 mm a 457 mm. Uma outra variedade produzia sementes
verdes, e outras ainda produziam sementes amarelas. Mendel aproveitou estas características
contrastantes para determinar como as características das plantas são herdadas. Seu enfoque nestas
diferenças singulares entre as linhagens de ervilhas lhe permitiu estudar a herança de uma
característica de cada vez — por exemplo, altura da planta. Outros biólogos tinham tentado seguir
simultaneamente a herança de muitas características, mas como os resultados de tais experimentos
eram complexos, eles foram incapazes de descobrir os princípios fundamentais sobre a
hereditariedade. Mendel teve sucesso onde estes biólogos haviam falhado porque enfocou sua
atenção em diferenças contrastantes entre as plantas que eram de outro modo iguais, alta
versus baixa, sementes verdes versus amarelas, e assim em diante. Além disso, ele fez registros
cuidadosos dos experimentos feitos.
Cruzamentos Monoíbridos: Os Princípios da Dominância e da Segregação
Em um experimento, Mendel cruzou plantas altas com baixas para investigar como a altura
era herdada (Figura 1).
Figura 1. Espécie de ervilha
utilizada por Mendel.
11
Ele removeu cuidadosamente as anteras de uma variedade antes que seu pólen tivesse
amadurecido, e em seguida aplicou pólen de outra variedade ao estigma, um órgão pegajoso na
parte superior do pistilo, que leva ao ovário. As sementes que resultaram destas fertilizações
cruzadas brotaram no ano seguinte, dando híbridos uniformemente altos. Mendel obteve plantas
altas independente do modo como o cruzamento foi feito (planta masculina alta com feminina anã
ou masculina anã com feminina alta). Assim, os cruzamentos recíprocos deram os mesmos
resultados. Mais significativamente, entretanto, Mendel notou que a característica anã parecia ter
desaparecido na prole do cruzamento, pois todas as plantas híbridas eram altas.
Para
explorar a constituição hereditária destes híbridos altos, Mendel permitiu que houvesse
autofecundação - o curso natural de eventos nas ervilhas. Quando ele examinou a prole, observou
que elas consistiam tanto em plantas altas quanto anãs. De fato, entre l .064 indivíduos da prole que
Mendel cultivou em seu jardim, 787 eram altos e 277 eram anões, uma proporção de
aproximadamente 3:1.
Mendel se espantou com o reaparecimento da característica anã. Claramente, os híbridos que
ele havia obtido cruzando as variedades alta e baixa tinham a habilidade de produzir uma prole anã,
muito embora fossem altos. Mendel deduziu que estes híbridos levavam um fator genético
latente para anã, que foi mascarado pela expressão de outro fator para alta. Ele disse que o fator
latente era recessivo e que o fator expresso era dominante. Deduziu também que estes fatores
recessivo e dominante se separaram um do outro quando as plantas híbridas se reproduziram. De
que outro modo ele poderia explicar o reaparecimento da característica anã na geração seguinte?
Mendel fez experimentos semelhantes para estudar a herança de seis outras características:
textura da semente, cor da semente, forma da vagem, cor da flor e posição da flor (Quadro .1).
Quadro 1. Demostração das características estudadas por Mendel.
Em cada experimento, chamado cruzamento monoíbrido, pois só uma característica estava
sendo estudada, Mendel observou que apenas uma de duas características contrastantes aparecia nos
híbridos, e que quando estes híbridos eram autofecundados produziam dois tipos de prole, cada uma
semelhante a uma das plantas no cruzamento original. Além disso, observou que esta prole aparecia
12
consistentemente em uma proporção de 3:1. Assim, cada característica que Mendel estudou parecia
ser controlada por um fator herdável que existia em duas formas, uma dominante e outra
recessiva. Estes fatores hoje são chamados de genes, uma palavra criada por um agricultor
dinamarquês chamado Wilhelm Johannsen em 1909; suas formas dominante e recessiva são
chamadas alelos, da palavra grega que significa "de outro tipo".
As relações numéricas regulares que Mendel observou nestes cruzamentos o levaram a outra
conclusão importante: a de que os genes existem aos pares. Mendel propôs que cada uma das
linhagens parentais que ele usou em seus experimentos levava duas cópias idênticas de um gene —
na terminologia moderna, elas são diplóides e homozigotas. Entretanto, durante a produção de
gamelas, Mendel propôs que estas duas cópias são reduzidas a uma; isto é, os gametas que emergem
da meiose levam uma só cópia de um gene — na terminologia moderna, eles são haplóides.
Mendel reconheceu que o número de genes diplóides seria restaurado quando o
espermatozóide e o ovócito se unissem para formar um zigoto. Além disso, ele compreendeu que se
o pólen e o ovócito viessem de plantas geneticamente diferentes, como em seus cruzamentos, o
zigoto híbrido herdaria dois alelos diferentes, um da mãe e um do pai. Diz-se que tal prole é
heterozigota. Mendel percebeu que alelos diferentes que estão presentes em um heterozigoto
devem coexistir, muito embora um seja dominante e o outro recessivo, e que cada um destes alelos
teria uma chance igual de ir para um gamela quando o heterozigoto se reproduzisse. Além disso, ele
percebeu que a fertilização aleatória com uma população mista de gamelas — metade com o alelo
dominante e metade com o alelo recessivo — produziria alguns zigolos nos quais ambos os alelos
eram recessivos. Assim, ele pôde explicar o reaparecimento da característica recessiva na prole de
plantas híbridas.
Mendel usou símbolos para representar os fatores hereditários que ele postulou — uma
conquista metodológica. Com símbolos, ele pôde descrever os fenómenos heredilários clara e precisamente, e pôde analisar os resullados dos cruzamentos matematicamente. Pôde até fazer
previsões sobre o resullado de cruzamentos futuros. Embora a prática de usar símbolos para analisar
problemas genéticos tenha sido muito refinada desde a época de Mendel, os princípios básicos
permanecem os mesmos. Os símbolos representam os genes (ou, mais precisamente, seus alelos) e
são manipulados de acordo com as regras da herança que Mendel descobriu. Essas manipulações
são a essência da análise genética formal. Como uma introdução a este assunto, consideremos a
represenlação simbólica do cruzamento entre ervilhas altas e anãs (Fig. 1).
13
As duas variedades puras, alta e anã, são homozigolas para alelos diferentes de um gene
controlador da altura da planta. O alelo para anã, sendo recessivo, é representado por uma letra
minúscula d; o alelo para alta, sendo dominante, é simbolizado pela mesma letra, mas maiúscula,
D. Em genética, a letra que é escolhida para indicar os alelos de um gene geralmente é tirada da
palavra que descreve a característica recessiva (d, de dwarfness). Assim, as linhagens de ervilhas
alta e anã são simbolizadas por DD e dd, respectivamente. Diz-se que a constituição alélica de cada
linhagem é o seu genótipo. Em oposição, diz que o aspecto de cada linhagem, isto é, a característica
alta ou anã é seu fenótipo.
Sendo as linhagens parentais, as plantas alta e anã de ervilha formam a geração P do
experimento. Sua prole híbrida é chamada de primeira geração filial, ou F1. Como cada genitor
contribui igualmente para sua prole, o genótipo das plantas de F1 deve ser Dd; isto é, elas são
heterozigotas para os alelos do gene que controla a altura da planta. Seu fenótipo, entretanto, é o
mesmo que o da linhagem parental DD, pois D é dominante em relação a d. Durante a meiose, estas
plantas de F, produzem dois tipos de gametas, D e d, em iguais proporções. Nenhum dos alelos é
alterado por ter coexistido no genótipo heterozigoto; em vez disso, eles se separam, ou se segregam,
um do outro durante a formação de gametas. Este processo de segregação de alelos é talvez a mais
importante descoberta de Mendel.
Na autofertilização, os dois tipos de gametas produzidos pelos heterozigotos podem se unir
de todos os modos possíveis. Assim, eles produzem quatro tipos de zigotos: DD (a contribuicão do
ovócito em geral é escrita primeiro), Dd, dD e dd. Entretanto, devido à dominância, três destes
genótipos têm o mesmo fenótipo. Assim, na geração seguinte, chamada de F2, as plantas são altas
ou anãs, em uma proporção de 3: l.
Mendel levou esta análise um pouco mais adiante. As plantas de F2 foram autofecundadas
para produzir uma F3. Todas as plantas anãs F2 produziram apenas prole anã, demonstrando que
eram homozigotas para o alelo d, mas as plantas altas de F, compreendiam duas categorias.
Aproximadamente um terço delas só produzia prole alta, enquanto os outros dois terços produziam
uma mistura de prole alta e anã. Mendel concluiu, corretamente que o terço que era "puro" era de
homozigotos DD, e os dois terços que se segregavam eram heterozigotos Dd. Estas proporções, 1/3
e 2/3, eram exatamente o que sua análise previu, pois entre as plantas altas de F2, os genótipos DD e
Dd ocorrem em uma proporção de 1:2.
Resumimos a análise feita por Mendel deste e de outros cruzamentos monoíbridos citando dois
princípios básicos que ele descobriu:
14
1. O Princípio da Dominância: Em um heterozigoto, um alelo pode encobrir a presença de outro.
Este princípio é relativo ao funcionamento genético. Alguns alelos evidentemente controlam o
fenótipo, mesmo quando estão presentes em uma única cópia. Consideraremos a explicação
fisiológica para este fenómeno em capítulos posteriores.
2. O Princípio da Segregação: Em um heterozigoto, dois alelos diferentes se segregam um do
outro durante a formação de gametas. Este princípio refere-se à transmissão genética. Um alelo é
transmitido fielmente para a geração seguinte, mesmo se estiver presente com um alelo diferente em
um heterozigoto. A base biológica deste fenômeno é o pareamento e subsequente separação de
cromossomos homólogos durante a meiose.
Cruzamentos Diíbridos: O Princípio da Distribuição Independente
Mendel também fez experimentos com plantas que diferiam em duas características (Fig. 2).
Ele cruzou plantas que produziam sementes amarelas e lisas com plantas que produziam sementes
verdes e rugosas. O propósito do experimento era ver se as duas características, cor e textura, eram
herdadas independentemente. Como as sementes de F, eram todas amarelas e lisas, os alelos para
estas duas características eram dominantes. Mendel cultivou plantas a partir destas sementes e
deixou que elas se autofecundassem. Então classificou as sementes de F2 e as contou por fenótipo.
As quatro classes fenotípicas na F2 representavam todas as combinações possíveis das
características de cor e textura. Duas classes, amarela lisa e verde rugosa, se assemelhavam às
linhagens parentais. As outras duas, verde lisa e amarela rugosa, mostravam novas combinações de
características. Mendel notou que as quatro classes apareciam em uma proporção de
aproximadamente 9 amarelas, lisas; 3 verdes, lisas; 3 amarelas, rugosas, e l verde, rugosa (Fig. 2).
Para sua mente perspicaz, estas relações numéricas sugeriram uma explicação simples: Cada
característica era controlada por um gene diferente que segregava dois alelos, e os dois genes eram
herdados independentemente.
Figura 2. Cruzamentos de Mendel
analisando duas características.
15
Estudemos os resultados deste cruzamento de dois fatores, ou cruzamento diíbrido, usando
os métodos de Mendel. Representaremos cada gene por uma letra, usando a minúscula para o alelo
recessivo e a maiúscula para o dominante (Fig.3). Para o gene de cor da semente, os dois alelos são
g (para verde) e G (para amarelo), e para a textura da semente, elas são w (para rugosa [wrinkled]) e
W (para lisa). As linhagens parentais, que eram puras, devem ter sido duplamente homozigotas; as
plantas amarelas, lisas eram GG WW, e as plantas verdes rugosas eram gg ww. Tais genótipos de
dois tipos são habitualmente escritos por pares separados de alelos com um espaço.
Os gametas haplóides produzidos por uma planta diplóide contêm uma cópia de cada gene. Os
gametas de plantas GG WW contêm, portanto, uma cópia do gene para a cor da semente (o alelo G)
e uma cópia para a textura da semente (o alelo W). Tais gametas são simbolizados por G W. Por um
raciocínio semelhante, os gametas das plantas gg ww são escritos g w. A fertilização cruzada destes
dois tipos de gametas produz híbridos de F, que são duplamente heterozigotos, simbolizados por Gg
Ww, e seu fenótipo amarelo liso indica que os alelos G e W são dominantes.
O Princípio da Segregação prevê que os híbridos de f! produzirão quatro genótipos
gaméticos diferentes: (1) G W, (2) G w, (3) g W e (4) g w. Se cada gene segrega seus alelos
independentemente, estes quatro tipos serão igualmente frequentes, isto é, cada um será 25 por
cento do total. Nesta suposição, a autofe-cundação na F, produzirá uma disposição de 16 genótipos
zigóticos igualmente frequentes. Obtemos a disposição zigótica combinando sistematicamente os
gametas, como mostrado na Fig. 3. Obtemos então os fenótipos destes genótipos de F2 observando
que G e W são alelos dominantes. Existem quatro fenótipos distinguíveis, com frequências relativas
indicadas pelo número de posições ocupadas na disposição. Para frequências absolutas, dividimos
cada número pelo total, 16:
Esta análise se baseia em duas suposições: (1) a de que cada gene segrega seus alelos e (2) a
de que estas segregações são independentes uma da outra. A segunda suposição significa que não há
conexão ou ligação entre os eventos de segregação dos dois genes. Por exemplo, um gamela que
recebe W pela segregação do gene de textura tem a mesma probabilidade de receber G que de
receber g pela segregação do gene de cor.
16
Figura
3.
Representação
simbólica dos resultados de um
cruzamento de Mendel.
Os dados experimentais se ajustam às previsões de nossa análise? A Fig. 4 compara as
frequências previstas e as observadas dos quatro fenótipos de F2 de dois modos — por proporções e
pelas frequências numéricas. Para as frequências numéricas, calculamos os números previstos
multiplicando a proporção prevista pelo número total de sementes de F2 examinadas. Com ambos os
métodos, obviamente há uma boa concordância entre as observações e as previsões. Portanto, as
suposições nas quais baseamos nossa análise — segregação independente dos genes de cor da
semente e da textura da semente — estão de acordo com os dados observados.
Mendel conduziu experimentos semelhantes com outras combinações de características, e em cada
caso observou que os genes se segregavam independentemente. Os resultados destes experimentos
o levaram a um terceiro princípio importante:
Figura 4. Comparação entre os resultados
esperados e obtidos na F2.
3. O Princípio da Distribuição Independente: Os alelos de genes diferentes se segregam, ou,
como às vezes dizemos, se distribuem independentemente uns dos outros. Este princípio é outra
17
regra da transmissão genética, baseada, como veremos no Cap. 6, no comportamento de pares
diferentes de cromossomos durante a meiose. Entretanto, nem todos os genes estão sujeitos ao
Princípio da Distribuição Independente.
Pontos Importantes: Os experimentos de Mendel estabeleceram três princípios genéticos básicos: (1) Alguns
alelos são dominantes, outros recessivos. (2) Durante a formação de gamelas, alelos diferentes se segregam uns
dos outros. (3) Genes diferentes se distribuem independentemente.
APLICAÇÕES DOS PRINCÍPIOS DE MENDEL
Se a base genética de uma característica for conhecida, os princípios de Mendel podem ser
usados para prever o resultado dos cruzamentos. Existem três procedimentos gerais, dois baseados
na numeração sistemática de todos os genótipos zigóticos ou fenótipos, e um que se baseia em
matemática.
O Método do Quadrado de Punnett
Para situações que envolvam um ou dois genes, é possível escrever todos os gametas e
combiná-los sistematicamente para gerar uma gama de genótipos zigóticos. Uma vez que eles
tenham sido obtidos, o Princípio da Dominância pode ser usado para determinar os fenótipos
associados. Este procedimento, chamado de método do quadrado de Punnett em homenagem ao
geneticista britânico R. C. Punnett, é um modo direto de se prever o resultado dos cruzamentos. Nós
o usamos para analisar o resultado zigótico do cruzamento com os híbridos de F, amarelos lisos, de
Mendel, um tipo de cruzamento comumente chamado de intercruzamento (Fig. 5). Entretanto, em
situações mais complicadas, como as que envolvem mais de dois genes, o método do quadrado de
Punnett não é recomendado.
O Método da Linha Bifurcada
Um outro procedimento para prever o resultado de um cruzamento envolvendo dois ou mais
genes é o método da linha bifurcada. Entretanto, em lugar de enumerar a prole em um quadrado
combinando sistematicamente os gamelas, os indicamos no diagrama com linhas ramificadas.
Como exemplo, consideremos um intercruzamento entre ervilhas que são heterozigotas para três
genes de segregação independente, um controlando a altura da planta, um controlando a cor da
semente, e um controlando a textura. Este é um cruzamento triíbrido — Dd Gg Ww X Dd Gg Ww —
que pode ser fracionado em três cruzamentos monoíbridos — Dd x Dd, Gg X Gg, e Ww x Ww —
pois todos os genes se segregam independentemente.
18
Para cada gene, esperamos que o fenótipo apareça em uma proporção de 3:1. Assim, por
exemplo, Dd X Dd produzirá uma proporção de 3 plantas altas: l planta anã. Usando o método da
linha bifurcada (Fig. 5), podemos combinar estas proporções separadas em uma proporção
fenotípica geral para a prole do cruzamento.
Figura 5. Método da linha
bifurcada.
Também podemos usar este método para analisar os resultados de um cruzamento entre
indivíduos multiplamente heterozigotos e multiplamente homozigotos. Este tipo de cruzamento é
chamado de cruzamento-teste. Por exemplo, se plantas de ervilhas Dd Gg Ww forem cruzadas com
outras dd gg ww, podemos prever os fenótipos da prole notando que cada um dos três genes no
genitor heterozigoto segrega alelos dominantes e recessivos em uma proporção de l:l, e que o
genitor homozigoto transmite apenas alelos recessivos destes genes. Assim, os genótipos, e
finalmente os fenótipos, da prole deste cruzamento dependem de quais alelos são transmitidos pelo
genitor heterozigoto (Fig. 6).
Figura 6. Método da linha
bifurcada
envolvendo
o
cruzamento-teste.
O Método da Probabilidade
Um método alternativo ao quadrado de Punnett e ao da linha bifurcada — também mais rápido — é
baseado no princípio da probabilidade (veja Enfoque Técnico: As Regras da Multiplicação e da
Adição da Probabilidade). A segregação mendeliana é como um cara ou coroa; quando o
heterozigoto produz gamelas, metade contém um alelo e metade contém o outro. Se dois
19
heterozigotos segregantes são cruzados, seus gamelas são combinados aleatoriamente, produzindo
os genótipos zigólicos (Fig. 7). Suponhamos que o cruzamento seja Aa x Aa. A chance de um zigolo
ser AA é simplesmente a probabilidade de que cada um gamela que se une contenha A, ou (1/2) x
(1/2) = (1/4), pois os dois gamelas são produzidos independentemente. A chance de um homozigolo
aã lambem é de 1/4. Enlrelanto, a chance de um heterozigoto é 1/2, pois existem dois modos de
criar um helerozigoto - A pode vir de um ovócito ou vir de um espermatozóide, ou vice-versa.
Figura 7. Intercruzamento com o quadrado
de Punnett.
Como cada um desses eventos tem uma chance de um para quatro de ocorrer, a probabilidade lolal
de que uma prole seja helerozigota é (1/4) + (1/4) = (1/2). Obtemos, portanto, a seguinte
probabilidade de distribuição dos genólipos do cruzamento Aa x Aa:
Concluímos que (1/4) + (1/2) = (3/4) da prole terá o fenótipo dominante, e 1/4 terá o
recessivo.
Para tal situação simples, o uso do método da probabilidade pode parecer desnecessário.
Entretanto, em situações mais complicadas, é claramente o enfoque mais prático para prever o
resultado de cruzamentos. Considere, por exemplo, um cruzamento entre planlas heterozigolas para
genes diferentes, cada um se segregando independentemente. Que fração da prole será homozigota
para todos os quatro alelos recessivos? Para responder a esla pergunta, consideramos os genes um
de cada vez. Para o primeiro gene, a fração da prole que será homozigota recessiva é 1/4, como será
para o segundo, terceiro e quarto genes. Portanto, pelo Princípio da Distribuição Independente, a
fração da prole que será de homozígotos quádruplos recessivos é (1/4) X (1/4) X (1/4) X (1/4) =
(1/256). Certamente, o uso do método da probabilidade é um enfoque melhor que um diagrama do
quadrado de Punnett com 256 partes!
20
Consideremos agora uma questão ainda mais difícil. Que fração da prole será homozigota
para todos os quatro genes? Antes de calcular qualquer probabilidade, devemos primeiro decidir
que genótipos satisfazem a questão. Para cada gene existem dois tipos de homozigotos, o dominante
e o recessivo, e juntos eles constituem metade da prole. A fração da prole que será homozigota para
todos os quatro genes será portanto (1/2) X (1/2) x (1/2) X (1/2) = (1/16).
Para entender todo o poder do método da probabilidade, precisamos considerar mais uma
questão. Suponha que o cruzamento é Aa Bb x Aa Bb e que desejamos saber que fração da prole
apresentará o fenótipo recessivo para pelo menos um gene (Fig. 3.9). Três tipos de genótipos
satisfariam esta condição: (l) A-bb (o traço representa A ou a), (2) aa B- e (3) aa bb. A resposta a
esta pergunta deve ser a soma das probabilidades que correspondem a cada um destes genótipos. A
probabilidade para A- bb é (3/4) X (1/4) = (3/16), a para aa B- é (1/4) x (3/4) = (3/16), e a para aa
bb é (1/4) X (1/4) = (1/16). Somando, temos a resposta, que é 7/16.
Figura 8. Aplicação dos métodos das probabilidades
a um cruzamento envolvendo dois genes.
Pontos Importantes: O resultado de um cruzamento pode ser previsto pela enumeração sistemática
dos genótipos em um quadrado de Punnett. Entretanto, quando estão envolvidos mais de dois genes, o
método da linha bifurcada ou o da probabilidade são usados para prever o resultado de um
cruzamento.
FORMULAÇÃO E TESTE DAS HIPÓTESES GENÉTICAS
Uma investigação científica sempre começa com observações de um fenómeno natural. As
observações levam a ideias ou perguntas sobre os fenômenos, e ambas são mais amplamente
exploradas fazendo-se outras observações ou experimentos. Uma ideia científica bem formulada é
chamada de uma hipótese. Os dados colhidos de observações ou de experimentação possibilitam
que os cientistas testem hipóteses, isto é, determinem se uma hipótese em particular deve ser aceita
ou rejeitada.
21
Como um exemplo de teste de uma hipótese em genética, consideremos a herança da cor da
flor na boca-de-leão, Antirrhinum majus, uma planta popular de jardim. Duas linhagens puras foram
obtidas de um estoque de laboratório, uma com flores vermelho-escuras e uma com flores brancas.
Estas diferenças de cor têm uma base genética? Para responder a esta pergunta, duas linhagens
devem ser cruzadas para produzir híbridos de F1, todos com flores rosa. Quando os híbridos de F1,
foram intercruzados, eles produziram três tipos de plantas de F2: vermelha (62), rosa (131) e branca
(57), com seus números mostrados nos parênteses. Como podemos explicar os dados?
Podemos supor que a cor da flor é controlada por um único gene com dois alelos W (para
vermelho) e w (para branco), e que as flores dos heterozigotos Ww são rosa porque W é apenas
parcialmente dominante em relação a w. De acordo com esta hipótese, a geração P seria WW
(vermelha) X ww (branca), produzindo híbridos de F, que seriam Ww (rosa), as quais, quando
intercruzadas, dariam uma prole F2 WW (vermelha), Ww (rosa), e ww (branca) em uma proporção
l :2: l. Os números reais parecem confirmar, dando crédito a esta hipótese.
O TESTE DO QUI-QUADRADO
Podemos perguntar se os dados de fato apoiam uma determinada hipótese. Esta pergunta é
critica, pois o valor de uma hipótese depende de sua habilidade em explicar os dados. Uma hipótese
que não se ajusta precisa ser modificada ou descartada em favor de algo melhor. Um procedimento
para testar a correspondência entre as previsões de uma hipótese e os dados reais usa uma estatística
chamada qui-quadrado (x2). Uma estatística é um número calculado a partir de dados - por
exemplo, a média de um conjunto de valores examinados. A estatística do x2 permite que um
pesquisador compare dados, tais como os números que obtemos de um experimento de cruzamento,
com seus valores previstos. Se a comparação for desfavorável, isto é, os dados não estiverem de
acordo com os valores previstos, a estatística do x2 excederá um número crítico, e a hipótese
genética será rejeitada. Se a estatística do x2 estiver abaixo deste número, a hipótese será aceita. A
estatística do x2, portanto, reduz o teste da hipótese a um procedimento simples e objetivo.
Como exemplo, consideremos os cruzamentos da boca-de-leão já descritos. Os dados de F2,
parecem ser coerentes com a hipótese de que um único gene está segregando dois alelos. Entretanto; para avaliar objetivamente esta hipótese, precisamos comparar os dados com seus valores
previstos. A Fig. 9 ilustra os cálculos.
22
Figura 9. Comparação dos resultados
observados e esperados e cálculos de quiquadrado.
O procedimento é direto. Para cada classe fenotípica, calculamos o número esperado de
prole multiplicando a proporção mendeliana e o tamanho total da amostra. Depois calculamos a
diferença entre os números observados e esperados e elevamos ao quadrado estas diferenças para
eliminar os efeitos canceladores de valores positivos e negativos. Após dividir cada diferença ao
quadrado pelo número correspondente esperado de prole, somamos todos os termos e comparamos
o x2 resultante com a distribuição de valores do x2 (Fig. 10).
Figura 10. Distribuição de uma
estatística de x2.
A distribuição de valores do x2, que é estabelecida por teoria estatística, mostra com que
frequência o x2 excederá um valor particular apenas por acaso. Os estatísticos recomendam estabelecer como limiar o limite de 5 por cento da distribuição. Se a hipótese for correta, o x2 estatístico
excederá este valor crítico de 5 por cento. Entretanto, se a hipótese for incorreta, haverá uma
chance maior de que o x2 exceda o valor crítico. Uma hipótese incorreta mais provavelmente
produzirá grandes diferenças entre as observações e expectativas. Quando tais diferenças grandes
forem elevadas ao quadrado, elas produzirão um grande válor do x2 estatístico, um situado à direita
23
da escala teórica. E costume rejeitar a hipótese se o x2 exceder o valor crítico. Assim, se a hipótese
for verdadeira, há uma chance de 5 por cento de erradamente ser rejeitada.
Voltando ao exemplo: o x2 calculado, 0,776, deve ser comparado ao valor crítico de uma
distribuição teórica. Ocorre, porém, que existem muitas destas distribuições, e para selecionar a
apropriada precisamos conhecer o grau de liberdade associado ao x2 estatístico. Este indicador das
distribuições de x2 é determinado subtraindo-se um do número de classes fenotípicas. Neste
exemplo, o número do grau de liberdade é 3 - l = 2. Podemos agora comparar a estatística do x 2
com o valor crítico da distribuição teórica com 2 graus de liberdade (veja o Quadro 2 para uma lista
de valores críticos). Como a estatística calculada, 0,776, é menor que o valor crítico, 5,991, a
hipótese de um gene segregando dois alelos não é rejeitada. Concluímos que esta hipótese é uma
explicação adequada para os dados.
Um problema desenvolvido ao final do capítulo mostra o que acontece quando o x
2
estatístico é maior que o valor crítico. Outros problemas criam oportunidades para usar o x
2
estatístico.
Pontos Importantes: O teste do qui-quadrado é um modo simples de se avaliar se as previsões
de uma hipótese genética concordam com os dados de um experimento.
PRINCÍPIOS MENDELIANOS EM GENÉTICA HUMANA
A aplicação dos princípios mendelianos à genética humana começou logo após a
redescoberta da publicação de Mendel em 1900. Entretanto, como não é possível fazer reproduções
controladas com seres humanos, o progresso foi obviamente lento. A análise genética da
hereditariedade humana depende de registros familiares, que em geral são incompletos. Além disso,
os seres humanos, ao contrário de organismos experimentais, não geram muitos descendentes,
dificultando discernir as proporções mendelianas. O erro de paternidade é outro problema em genética humana, introduzindo um elemento de confusão aos dados. O tempo também é um fator, pois
algumas condições genéticas não se manifestam até que uma pessoa atinja a meia-idade. Por todos
estes motivos, a análise genética humana tem sido uma tarefa difícil. Entretanto, a motivação para
compreender a hereditariedade humana tem sido muito forte, e hoje, a despeito de todos os
obstáculos, aprendemos sobre milhares de genes humanos. O Quadro 2 mostra algumas das
condições que eles controlam.
24
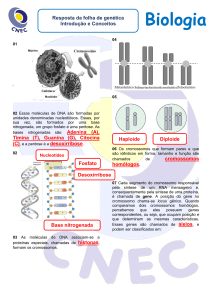
HEREDOGRAMAS
Os heredogramas (pedigrees) são diagramas que mostram o parentesco entre os membros
de uma família (Fig. 11). É costume representar os homens por quadrados e as mulheres por
círculos. Uma linha horizontal juntando um círculo e um quadrado representa uma reprodução. A
prole da reprodução é mostrada abaixo dos genitores, começando com o que nasceu primeiro à
esquerda e continuando, por ordem de nascimento, para a direita. As pessoas que têm uma condição
genética são indicadas em cores ou em preto. As gerações em um heredograma são geralmente
indicadas por algarismos romanos, e as pessoas em uma geração por arábicos após os romanos.
As características causadas por alelos dominantes são mais fáceis de identificar. Em geral, cada
pessoa que possui o alelo dominante manifesta a característica, possibilitando acompanhar a
transmissão deste alelo no heredograma (Fig. 11). Cada pessoa afetada deve ter pelo menos um
genitor afetado, a menos que, é claro, o alelo dominante tenha aparecido na família como resultado
de uma mutação nova, uma mudança no gene. Entretanto, a frequência de mutações novas é muito
baixa, da ordem de um em um milhão. Conseqüentemente, o surgimento espontâneo de uma
condição dominante é um evento extremamente raro. As características dominantes que estão
associadas à viabilidade reduzida ou à fertilidade nunca se tornam frequentes em uma população.
Assim, a maioria das pessoas que apresentam tais características são heterozigotas para o alelo
dominante. Caso seu cônjuge não tenha a característica, metade de seus filhos podem herdar a
condição.
Figura 11. (a) Convenções de
heredograma. (b) Herança de uma
característica dominante. A característica
aparece em cada geração, (c ) Herança de
uma característica recessiva. Os dois
afetados são descendentes de parentes.
25
As características recessivas não são fáceis de identificar porque podem ocorrer em pessoas
cujos genitores não são afetados. Às vezes são necessárias várias gerações de dados de heredograma
para acompanhar a transmissão de um alelo recessivo. Entretanto, um grande número de
características recessivas têm sido observadas em seres humanos - na última contagem, mais de
4.000. As características recessivas raras são mais prováveis de aparecer em um heredograma
quando os cônjuges são aparentados - por exemplo, quando são primos em primeiro grau. Este
aumento de incidência ocorre porque os genitores compartilham alelos em virtude de algum
ancestral comum. Os irmãos compartilham metade de seus alelos, meio-irmãos um quarto de seus
alelos, e primos em primeiro grau um oitavo de seus alelos. Assim, quando estes parentes se
reproduzem, eles têm um risco maior de ter um filho que seja homozigoto para um alelo recessivo
em particular do que os genitores não aparentados. Muitos dos estudos clássicos em genética
humana se basearam na análise de reproduções entre parentes, principalmente primos em primeiro
grau.
SEGREGAÇÃO MENDELIANA EM FAMÍLIAS HUMANAS
Nas famílias humanas, o número de filhos produzidos por um casal é tipicamente pequeno.
Hoje, nos Estados Unidos, a média está ao redor de dois. Nos países em desenvolvimento, é de seis
a sete. Tais números não fornecem nada estatisticamente tão poderoso quanto os experimentos de
Mendel com ervilhas. Conseqüentemente, as proporções fenotípicas nas famílias humanas em geral
se desviam significativamente de suas expectativas mendelianas.
Como exemplo, consideremos um casal em que ambos sejam heterozigotos para um alelo recessivo
que, em homozigose, cause fibrose cística. Se o casal tiver quatro filhos, serão esperados três
26
normais e um afetado pela fibrose cística? A resposta é não. Embora este possa ser um resultado,
não é o único. Existem, de fato, cinco possibilidades distintas:
1. Quatro normais, nenhum afetado.
2. Três normais, um afetado.
3. Dois normais, dois afetados.
4. Um normal, três afetados.
5. Nenhum normal, quatro afetados.
Intuitivamente, o segundo resultado parece ser o mais provável, pois está de acordo com a
proporção mendeliana de 3:1. Podemos calcular a probabilidade deste resultado, e de cada um dos
outros, usando os princípios de Mendel e tratando cada nascimento como um evento independente
(Fig. 12).
Figura 12. Distribuição da probabilidade de famílias com quatro filhos segregando uma característica recessiva.
Para um determinado nascimento, a chance de a criança ser normal é de 3/4. A
probabilidade de todos os quatro filhos serem normais é, portanto, de (3/4) X (3/4) X (3/4) X (3/4) =
(3/4)4 = 81/256. De modo semelhante, a chance de uma determinada criança ser afetada é de 1/4.
Assim, a probabilidade de todos os quatro serem afetados é (1/4)4 = 1/256. Para encontrar as
probabilidades dos outros três resultados, precisamos reconhecer que cada um de fato representa
uma coleção de eventos distintos. O resultado de três filhos normais e um afetado, por exemplo,
compreende quatro eventos distintos. Se simbolizarmos por N uma criança normal e por A uma
criança afetada, e se escrevermos as crianças em sua ordem de nascimento, podemos representar
estes eventos como:
NNNA, NNAN, NANN e ANNN
Como cada uma tem uma probabilidade de (3/4)3 X (1/4), a probabilidade total para três
crianças normais e uma afetada, independente da ordem de nascimento, é de 4 X (3/4)3 X (1/4). O
27
coeficiente 4 é o número de modos pêlos quais três podem ser normais e uma ser afetada em uma
família com quatro filhos. De modo semelhante, a probabilidade para dois filhos normais e dois
afetados é de 6 X (3/4)2 X (1/4)2, pois neste caso existem seis eventos distintos. A probabilidade
para uma criança normal e três afetadas é de 4 X (3/4) X (1/4)3, pois neste caso existem quatro
eventos distintos. A Fig. 3.14 resume os cálculos na fornia de uma distribuição de probabilidade.
Como esperado, três filhos normais e um afetado é o resultado mais provável (probabilidade
108/256). Entretanto, este resultado não é esperado na maior parte do tempo, pois os outros quatro
resultados têm uma probabilidade combinada de 148/256.0 Enfoque Técnico: Probabilidades
Binomiais generaliza este procedimento para outras situações nas quais as crianças caem em duas
possíveis classes fenotípicas. Como existem apenas duas classes, as probabilidades associadas aos
resultados são chamadas probabilidades binomiais.
CONSULTA GENÉTICA
O diagnóstico das condições genéticas em geral é um processo difícil. Tipicamente, os
diagnósticos são feitos por médicos que foram treinados em genética. O estudo destas condições
requer uma grande quantidade de pesquisas cuidadosas, inclusive examinar os pacientes, entrevistar
os parentes e pesquisar estatísticas vitais de nascimentos, mortes e casamentos. Os dados acumulados fornecem a base para se definir clinicamente a condição e para determinar seu modo de
herança.
Os futuros pais podem querer saber se seus filhos correm risco de herdar uma determinada
condição, especialmente se outros membros da família foram afetados. É responsabilidade do
consultor genético avaliar tais riscos e explicá-los aos futuros pais. A avaliação do risco requer
familiaridade com probabilidade e estatística, aliada a um bom conhecimento de genética.
Como exemplo, consideremos um heredograma mostrando uma forma rara de nanismo (Fig. 13).
28
Figura 13. Representação de uma família com nanismo.
A condição se manifesta em todas menos na primeira geração, e todos os afetados, exceto o
primeiro, têm um genitor afetado. Este padrão indica fortemente que a condição é causada por um
alelo dominante. A mulher anã na geração II provavelmente representa uma mutação nova na
população.
O aspecto da consulta surge na geração V. Qual a chance de um homem anão produzir uma
criança anã? Para responder a esta questão precisamos conhecer os genótipos dos futuros genitores.
Como a condição é causada por um alelo dominante, a mãe deve ser homozigota para o alelo
normal (recessivo) (dd) e o pai deve ser heterozigoto para o alelo de nanismo (Dd). A chance de o
casal ter um filho anão é portanto de 1/2.
Como outro exemplo, considere a situação mostrada na Fig. 3.16. Um casal, indicado por R e S,
está preocupado com a possibilidade de seu filho (T) ter albinismo (fig.14), uma condição autossômica recessiva caracterizada pela ausência completa do pigmento melanina na pele, nos olhos e nos
cabelos. S, o futuro pai, é um albino, e R, a futura mãe, tem dois irmãos albinos. Pareceria, portanto,
que o filho tem algum risco de nascer albino.
Figura 14. Albinismo em uma família.
29
O risco depende de dois fatores: (1) a probabilidade de R ser um portador heterozigoto do
alelo de albinismo (a), e (2) a probabilidade de ela transmitir este alelo para T se de fato for uma
portadora. S, que é obviamente homozigoto para o alelo do albinismo, deve transmitir este alelo
para sua prole.
Para determinar a primeira probabilidade, precisamos considerar os possíveis genótipos de R. Um
deles, de ela ser homozigota para o alelo recessivo (aa), é excluída porque sabemos que ela não é
albina. Entretanto, os outros dois genótipos, AA e Aa, continuam possibilidades distintas. Para
calcular as probabilidades associadas a cada um deles, notamos que ambos os genitores de R devem
ser heterozigotos, pois já tiveram dois filhos albinos. A reprodução que gerou R foi portanto Aa x
Aa, e de tal reprodução podemos esperar 2/3 da prole não-albina como sendo Aa e 1/3 como sendo
AÃ. Assim, a probabilidade de R ser portadora hete-rozigota do alelo do albinismo é de 2/3. Para
determinar a probabilidade de ela transmitir este alelo para seu filho, simplesmente observamos que
a estará presente em metade de seus gamelas. Em resumo, Risco de T ser aa = [Probabilidade de R
ser Aa] x [Probabilidade de R transmitir a, supondo que R seja Aa] = (2/3) X (1/2) = (1/3).
Os exemplos nas Figs. 3.15 e 3.16 ilustram simples situações de consulta genética na qual o
risco pode ser determinado com precisão. Em geral as circunstâncias são muito mais complicadas,
tornando a tarefa de avaliação do risco mais difícil. A responsabilidade do consultor genético é
analisar a informação do heredograma e determinar o risco tão precisamente quanto possível.
Pontos Importantes: Os heredogramas são usados para identificar características dominantes e recessivas em famílias
humanas. A análise de heredogramas permite que os consultores genéticos determinem a probabilidade de uma pessoa
herdar uma determinada característica.
30
RELEVANCIA DOS GENES E CROMOSSOMOS
CROMOSSOMOS
Os cromossomos foram descobertos na segunda metade do século XIX por um citologista alemão,
W. Waldeyer. As investigações subsequentes com organismos diferentes estabeleceram que os
cromossomos são característicos dos núcleos de todas as células. Eles são melhor vistos aplicando-se
corantes às células em divisão. Nesta fase, o material em um cromossomo está comprimido em um
pequeno volume, dando a aparência de um cilindro bem compactado. Durante a intérfase, entre as
multiplicações celulares, os cromossomos não são facilmente vistos, mesmo com os melhores corantes.
Os cromossomos interfásicos estão espalhados, formando finos filamentos distribuídos pelo núcleo.
Conseqüentemente, quando os corantes são aplicados, todo o núcleo é corado, e os cromossomos
individuais não podem ser identificados. Esta rede difusa de filamentos é chamada de cromatina.
Algumas regiões da cromatina coram-se mais fortemente que outras, sugerindo uma diferença
subjacente em organização. As regiões claras e escuras, respectivamente chamadas de
eucromatina (eu = verdadeiro, portanto, cromatina verdadeira) e a heterocromatina (hetero = diferente,
portanto, cromatina diferente), têm densidades diferentes de filamentos cromossômicos.
NÚMERO DE CROMOSSOMOS
Dentro de uma espécie, o número de cromossomos é quase sempre um múltiplo par de um número
básico. Nos seres humanos, por exemplo, o número básico é 23. Os ovócitos e espermatozóides têm este
número de cromossomos. A maioria dos outros tipos de células humanas tem o dobro (46), embora alguns
tipos, como as células do fígado, tenham quatro vezes (92) o número básico.
O número cromossômico haplóide, ou básico, (n) define um conjunto de cromossomos chamados
genoma haplóide. A maioria das células somáticas contém dois de cada cromossomo, sendo portanto
diplóides (2n). As células com quatro de cada cromossomo são tetraplóides (4n), as com oito de cada
são octaplóides (8n), e assim em diante.
O número básico de cromossomos varia entre as espécies. O número de cromossomos não está
relacionado ao tamanho nem à complexidade biológica de um organismo, e a maioria das espécies contém
entre 10 e 40 cromossomos em seus genomas (Quadro.3). O muntjac, um pequeno veado asiático, tem apenas três cromossomos em seu genoma, enquanto algumas espécies de samambaia têm muitas centenas.
31
CROMOSSOMOS SEXUAIS
Em algumas espécies animais, por exemplo, os gafanhotos, as fêmeas têm um cromossomo a
mais que os machos (Fig. 15). Este cromossomo extra, originalmente observado em outros insetos, é
chamado de cromossomo X. As fêmeas de outras espécies têm dois cromossomos X, e os machos têm
apenas um. Assim, as fêmeas são citologicamente XX e os machos são XO, onde o "O" indica a
ausência de um cromossomo. Durante a meiose na fêmea, os dois cromossomos X se pareiam e depois se
separam, produzindo ovócitos que contêm um único cromossomo X. Durante a meiose no macho, o
cromossomo X solitário move-se independentemente de todos os outros cromossomos e é incorporado
a metade dos espermatozóides. A outra metade não recebe nenhum cromossomo X. Assim, quando os
espermatozóides e ovócitos se unem, são produzidos dois tipos de zigotos: XX, que se desenvolvem em
fêmeas, e XO, que se desenvolvem em machos. Como cada um destes tipos é igualmente provável, o
mecanismo reprodutivo preserva uma proporção l: l de machos e fêmeas nestas espécies.
Em muitos outros animais, inclusive seres humanos, machos e fêmeas têm o mesmo número de
cromossomos (Fig.15). Esta igualdade numérica é devida à presença de um cromossomo no macho,
chamado cromossomo Y, que se pareia com o X durante a meiose. O cromossomo Y é morfologicamente distinguível do cromossomo X. Em humanos, por exemplo, o Y é muito mais curto que o X, e
seu centrômero está situado mais perto das pontas (Fig. 15). O material comum aos cromossomos
32
humanos X e Y é limitado, consistindo principalmente de curtos segmentos terminais. Durante a meiose
no macho, os cromossomos X e Y se separam um do outro, produzindo dois tipos de espermatozóides,
com X e com Y. As frequências dos dois tipos é aproximadamente igual. As fêmeas XX produzem
apenas um tipo de ovócito, que tem X. Se a fertilização ocorrer aleatoriamente, aproximadamente
metade dos zigotos terá XX e a outra metade terá XY, levando a uma proporção sexual de 1:1 na
concepção. Entretanto, em seres humanos, os espermatozóides portadores de Y têm uma vantagem de
fertilização, e a proporção sexual zigótica é de cerca de 1,3:1. Durante o desenvolvimento, o excesso de
machos é diminuído pela viabilidade diferencial dos embriões XX e XY, e ao nascimento os machos são
apenas um pouco mais numerosos que as fêmeas (proporção sexual de 1,07:1). Na idade reprodutiva, o
excesso de machos é essencialmente eliminado, e a proporção sexual é muito próxima de l: l.
Os cromossomos X e Y são chamados de cromossomos sexuais. Todos os outros cromossomos no
genoma são chamados de autossomos. Os cromossomos sexuais foram descobertos nos primeiros anos do
século XX pelo trabalho dos citologistas americanos C. E. McClung, N. M. Stevens, W. S. Sutton e E. B.
Wilson. Esta descoberta coincidiu muito com o surgimento do mendelismo e estimulou as pesquisas sobre as
possíveis relações entre os princípios de Mendel e o comportamento meiótico dos cromossomos.
Figura 15. Herança de
cromossomos sexuais em
animais.
Pontos Importantes: Os cromossomos emergem de uma rede difusa de fibras de cromatina durante a
multiplicação celular. As células somáticas diplóides têm o dobro dos cromossomos que os gametas
haplóides. Em algumas espécies, os cromossomos sexuais X e Y distinguem as células de machos e de
fêmeas — XY nos machos e XX nas fêmeas. Em outras espécies, a fêmea tem dois cromossomos X e
o macho tem um só X e nenhum Y.
TEORIA CROMOSSÔMICA DA HERANÇA
Em 1910, muitos biólogos suspeitaram de que os genes estavam situados em cromossomos, mas não
tinham prova definitiva. Os pesquisadores precisavam encontrar um gene que pudesse ser
inconfundivelmente ligado a um cromosspmo. Esta meta necessitava que o gene fosse definido por um alelo
mutante e que o cromossomo fosse morfologicamente distinguível. Além disso, o padrão de transmissão
33
gênica tinha que refletir o comportamento cromossômico durante a reprodução. Todos estes requisitos foram
atendidos quando o biólogo americano Thomas H. Morgan descobriu uma determinada mutação de cor de
olho na mosca das frutas, a Drosophila melanogaster. Morgan começou a experimentação com esta
espécie de mosca em 1909. Ela era idealmente adequada à pesquisa genética porque se reproduzia rápida e
prolifícamente e não era dispendiosa de se criar em laboratório. Além disso, ela só tem quatro pares de
cromossomos, sendo um par de cromossomos sexuais — XX na fêmea e XY no macho. Os cromossomos
X e Y eram morfologicamente dis-tinguíveis um do outro e de cada um dos autossomos. Através de
cuidadosos experimentos, Morgan foi capaz de mostrar que a mutação para cor de olhos era herdada
juntamente com o cromossomo X, sugerindo que um gene para cor de olhos estava fisicamente situado
neste cromossomo. Mais tarde, um de seus alunos, Calvin B. Bridges, obteve uma prova definitiva desta
Teoria Cromossômica da Herança.
Evidência Experimental Ligando a Herança de Genes aos Cromossomos
Os experimentos de Morgan começaram com a descoberta de uma mosca macho mutante que tinha
olhos brancos em vez dos olhos vermelhos das moscas tipo selvagem. Quando este macho foi cruzado com
fêmeas tipo selvagem, toda a prole teve olhos vermelhos, indicando que branco era recessivo em relação
a vermelho. Quando esta prole foi intercruzada uma com a outra, Morgan observou um padrão peculiar de
segregação: todas as filhas, mas apenas metade dos filhos, tinham olhos vermelhos. A outra metade dos
filhos tinha olhos brancos. Este padrão sugeriu que a herança da cor de olho estava ligada a cromossomos
sexuais. Morgan propôs que um gene para cor de olho estava presente no cromossomo X, mas não no Y,
e que os fenótipos branco e vermelho eram devidos a dois alelos diferentes, um alelo mutante indicado por w e
um alelo selvagem indicado por w+. A hipótese de Morgan está representada na Fig. 3. As fêmeas tipo
selvagem no primeiro cruzamento foram imaginadas como sendo homozigotas para o alelo w+. Seus
machos su-postamente tinham o alelo w mutante em seu cromossomo X e nenhum dos alelos em seu
cromossomo Y. Um organismo que tem apenas uma cópia de um gene é chamado de hemizij Entre a prole
do cruzamento, os filhos herdam um crome mo X de sua mãe e um cromossomo Y de seu pai. Como
herdado maternamente tem o alelo w+, estes filhos têm c vermelhos. As filhas, ao contrário, herdam um
cromosson de cada genitor, um X com w+ da mãe e um X com w do Entretanto, como w+ é dominante em
relação a w, estas fêr heterozigotas da F, também têm olhos vermelhos.
Quando os machos e fêmeas da F, são intercruzados, são duzidas quatro classes genotípicas, cada uma
representando combinação diferente de cromossomos sexuais. As moscas que são fêmeas, têm olhos
vermelhos porque pelo menos um í w+ está presente. As moscas XY, que são machos, têm o vermelhos ou
34
brancos, dependendo de qual cromossomo herdado das fêmeas heterozigotas de F1- A segregação dos los w
e w+ nestas fêmeas é portanto o motivo pelo qual me; dos machos da F2 têm olhos brancos.
Morgan fez experimentos adicionais para confirmar os mentos de sua hipótese. Em um deles (Fig.
16), ele cru fêmeas supostas heterozigotas para o gene de cor de olho ( machos mutantes de olhos brancos.
Como ele esperava, m de da prole de cada sexo tinha olhos brancos e metade tem olhos vermelhos. Em
outro experimento (Fig. 16), ele cruzou fêmeas de olhos brancos com machos de olhos vermelhos. Desta
vez, todas as filhas tinham olhos vermelhos, e todo os filhos tinham olhos brancos. Quando ele intercruzou
esta fêmea de, Morgan observou a segregação esperada: metade da pi de cada sexo tinha olhos brancos e
metade tinha olhos vermelhos. Assim, a hipótese de Morgan de que o gene para cor olho estava ligado ao
cromossomo X foi confirmada em tes experimentais adicionais.
Figura 16 O experimento de Morgan
estudando a herança de olhos brancos na
Drosophila. A transmissão da condição
mutante em associação ao sexo sugeriu que o
gene para cor de olho estava presente no
cromossomo X mas não no cromossomo Y.
CROMOSSOMOS COMO VEÍCULOS DE GENES
Morgan e seus alunos logo identificaram outros genes ligac ao X na Drosophila. Em cada caso,
experimentos simples cruzamentos demonstraram que mutações recessivas destes genes eram transmitidas
com o cromossomo X. À medida que evidências se acumularam, ficou claro que muitos genes estavavam
situados no cromossomo X. Entretanto, o grupo de pesquisa Morgan também identificou genes que não
estavam no cromossomo X. Estes genes seguiam o princípio mendeliano da segregação, mas não se
segregavam com o sexo, como o gene para cor de olho o fez. Morgan concluiu corretamente que tais genes
estavam situados em um dos três autossomos no gene Drosophila. Assim, cada cromossomo da
Drosophila contém um conjunto diferente de genes.
35
O laboratório de Morgan tentou então determinar as relações entre os genes em um cromossomo
em particular. Eles continuaram com a suposição de que os genes estavam postos de modo linear, uma
idéia inspirada pela evidencia citológica do cromossomo era um filamento longo e fino. Em apenas
alguns anos, os alunos de Morgan foram capazes de mostrar que os genes estavam de fato situados em
pontos diferentes, ou loci (da palavra latina para "lugar"), em uma estrutura linear (Fig. 17). O
laboratório de Morgan foi pioneiro nos método de construção de mapas genéticos e estabeleceu as
fundações para pesquisas subsequentes sobre a estrutura física dos cromossomos. Posteriormente a
estrutura linear dos cromossomos. Posteriormente, a linearidade dos cromossomos foi associada a
estrutura linear do DNA.
Estes primeiros estudos com Drosophila — principalmente o trabalho de Morgan e seus alunos
(Enfoque Histórico: Drosophila, T. H. Morgan e "A Sala das Moscas") — fortaleceram muito a visão de
que todos os genes estão situados em cromossomos e que os princípios de Mendel podiam ser explicados pelas
propriedades de transmissão dos cromossomos durante a reprodução. Esta ideia, chamada de Teoria
Cromossômica da Hereditariedade, é uma das mais importantes conquistas da biologia. Desde sua
formulação na primeira parte do século XX, a Teoria Cromossômica da Hereditariedade forneceu uma
estrutura unificadora para todos os estudos sobre a herança (fig. 18).
A) cruzamento entre uma fêmea heterozigotae
um macho mutante hemizigoto.
B) cruzamento entre uma fêmea mutante
homozigota e um macho selvagem
heterozigoto
Fig. 17 Testes experimentais da hipótese de Morgan de que o gene para cor de olho na Drosophila
é ligado ao X. Em cada experimento, a cor de olho é herdada juntamente com o cromossomo X.
Logo, os resultados destes cruzamentos apoiaram a hipótese de Morgan de que o gene para cor de
olho é ligado ao X.
36
Fig. 18. Um mapa de genes
no cromossomo X da
Drosophila.
Não-disjunção como Prova da Teoria Cromossômica
Morgan mostrou que um gene para cor de olho estava no cromossomo X da Drosophila
correlacionando a herança deste gene com a transmissão dos cromossomos X durante a reprodução.
Entretanto, como já dito antes, foi um de seus alunos, C. B. Bridges, quem deu a prova segura da teoria
Cromossômica, mostrando que as exceções às regras da herança também podiam ser explicadas pelo
comportamento cromossômico.
Bridges fez um dos experimentos de Morgan em uma escala maior. Ele cruzou fêmeas de
Drosophila de olhos brancos com machos de olhos vermelhos e examinou uma grande prole da F1. Embora,
como esperado, quase todas as moscas de F1 fossem fêmeas de olhos vermelhos ou machos de olhos
brancos, Bridges observou algumas moscas excepcionais — fêmeas de olhos brancos e machos de olhos
vermelhos. Ele cruzou estas exceções para determinar como elas podiam ter surgido. Os machos excepcionais
eram todos estéreis. Entretanto, as fêmeas excepcionais eram férteis, e quando cruzadas com machos
normais de olhos vermelhos produziam uma grande prole, incluindo um enorme número de filhas com
olhos brancos e filhos com olhos vermelhos. Assim, as fêmeas excepcionais da F1 embora raras, geravam
muita prole excepcional.
Bridges explicou estes resultados propondo que as moscas excepcionais da F1 eram o resultado do
comportamento anormal do cromossomo X durante a meiose nas fêmeas da geração P. Ordinariamente, os
cromossomos X nestas fêmeas deveriam se disjuntar, ou separar, um do outro durante a meio-se.
37
Ocasionalmente, entretanto, eles podiam falhar em se separar, produzindo um ovócito com dois
cromossomos X ou um sem nenhum cromossomo X. A fertilização de tais ovócitos anormais por
espermatozóides normais produziria zigotos com um número anormal de cromossomos sexuais. A Fig. 19
ilustra as possibilidades.
Se um ovócito com dois cromossomos X (em geral chamado diplo X; genótipo XwXw) for fertilizado
por um espermatozóide portador de Y, o zigoto será XWXWY. Como cada um dos cromossomos X neste
zigoto leva um alelo w mutante, a mosca resultante terá olhos brancos. Se um ovócito sem cromossomo X
(em geral chamado de X nulo) for fertilizado por um espermatozóide X (X+,), o zigoto será X+O. (Novamente,
"0" indica a ausência de um cromossomo). Como o único X neste zigoto leva um alelo w+, ele se
desenvolverá em uma mosca com olhos vermelhos. Bridges deduziu que as moscas XXY eram fêmeas e que
as moscas XO eram machos. As fêmeas excepcionais de olhos brancos que ele observou eram, portanto,
XWXWY, e os machos excepcionais de olhos vermelhos eramX+0. Bridges confirmou as constituições
cromossômicas destas moscas excepcionais por observação citológica direta. Como os animais XO eram
machos, Bridges concluiu que na Drosophila o cromossomo Y não tem nada a ver com a determinação do
fenótipo sexual. Entretanto, como os machos XO eram sempre estéreis, ele percebeu que este cromossomo
tinha que ser importante para o funcionamento sexual masculino.
Bridges reconheceu que a fertilização de ovócitos anormais por espermatozóídes normais podia
produzir dois tipos adicionais de zigotos: XWXWX+, surgindo da união de um ovócito duplo X e um
espermatozóide portador de X, e YO, surgindo da união de um ovócito X nulo e um espermatozóide com Y.
Os zigotos XWXWX+ se desenvolvem em fêmeas de olhos vermelhos, mas fracas e doentias. Estas "metafêmeas"
podem ser distinguidas das fêmeas XX por uma síndrome de anomalias anatômicas, incluindo asas
malformadas e abdomes corroídos. Gerações de geneticistas impropriamente as chamaram de "superfêmeas",
um termo criado por Bridges, muito embora não exista nada "supei nelas. Os zigotos YO são totalmente
inviáveis, isto é, morrem. Na Drosophila, como na maioria dos organismos com cromossomos sexuais, pelo
menos um cromossomo X é necessário para viabilidade.
A habilidade de Bridges para explicar a prole excepcional que surge destes cruzamentos mostrou
o poder da teoria cromossômica. Cada uma das exceções era devida a um comportamento cromossômico
anômalo durante a meiose. Bridges chamou a anomalia de não-disjunção, porque ela envolve uma falha
dos cromossomos em se separar durante uma das divisões meióticas. Esta falha pode resultar de um
movimento defeituoso dos cromossomos, de pareamento impreciso ou incompleto, ou de mau
funcionamento do centrômero. Pêlos dados de Bridges, é impossível especificar a causa exata.
Entretanto, Bridges notou que as fêmeas excepcionais XXY produzem uma alta frequência de prole
38
excepcional, supostamente porque seus cromossomos sexuais podem se separar de modos diferentes: os
cromossomos X podem se separar uns dos outros, ou os X dos Y. Neste último caso, um ovócito X diplo ou nulo é
produzido, porque o X que não se separa do Y está livre para se mover para um dos pólos durante a primeira meiose.
Quando fertilizado por um espermatozóide normal, estes ovócitos anormais produzirão zigotos excepcionais.
Fig. 19. A não disjunção do cromossomo X é responsável
pela prole excepcional que apareceu no experimento de
Bridges. Os ovócitos com não-disjunção que continham
ambos os cromossomos X ou nenhum comossomo X se
uniram com espermatozoides normais que continham um
cromossomo X ou um Y para produzir quatro tipos de
zigotos. Os zigotos XXY se desenvolvem em fêmeas de
olhos brancos, os zigotos XO se desenvolvem em machos
estéreis de olhos vermelhos, e os zigotos XXX e YO
A Base Cromossômica dos Princípios Mendelianos
da Segregação e da Distribuição Independente
morrem.
Mendel estabeleceu dois princípios da transmissão genética: (1) os alelos de um gene se
segregam um do outro, e (2) os alelos de genes diferentes se distribuem independentemente. O achado
de que os genes estão situados em cromossomos possibilitou explicar estes princípios (bem como
exce-ções a eles) em termos do comportamento meiótico dos cromossomos.
O princípio da segregação (Fig.20). Durante a primeira meiose, os cromossomos homólogos se pareiam.
Um dos homólogos vem da mãe, e o outro, do pai. Se a mãe era homozigota para um alelo, A, de um gene
neste cromossomo e o pai era homozigoto para um alelo diferente, a, do mesmo gene, a prole tem que ser
heterozigota, isto é, Aa. Na anáfase da primeira meiose, os cromossomos pareados se separam e se
deslocam para pólos opostos da célula. Um leva o alelo A e o outro o alelo a. Esta separação física dos
dois cromossomos segrega os alelos um do outro. Posteriormente, eles irão residir em células-filhas diferentes.
O princípio da segregação de Mendel é portanto baseado na separação dos cromossomos homólogos durante a
anáfase da primeira meiose.
39
40
Figura 20. Segregação de Mendel com a meiose.
O princípio da distribuição independente : O princípio da distribuição independente também é baseado nesta
separação anafásica. Para compreender a correlação, precisamos considerar os genes em dois pares
diferentes de cromossomos. Suponha que um heterozigoto Aa Bb foi produzido pelo cruzamento de uma
fêmea AA BB com um macho aa bb. Suponha, também, que os dois genes estão em cromossomos
diferentes. Durante a prófase da meiose I, os cromossomos com os alelos A e a se pareiam, como o fazem os
cromossomos com os alelos B e b. Na metáfase, os dois pares tomam posições no fuso meiótico em
preparação para a separação anafásica seguinte. Como existem dois pares de cromossomos, existem dois
alinhamentos metafásicos distintos:
A B
——
a b
ou
A b
——
a B
Cada um destes alinhamentos é igualmente provável. Aqui o espaço separa pares diferentes de
cromossomos e as barras separam os membros homólogos de cada par. Durante a anáfase, os alelos acima das
barras irão se mover para um pólo, e os alelos abaixo delas irão se mover para o outro. Quando ocorre a
disjunção, há portanto 50% de chance de que os alelos A e B se movam juntos para o mesmo pólo e uma
chance de 50% de que se movam para pólos opostos. Analogamente, há uma chance de 50% de que os alelos
a e b se movam para o mesmo pólo e de 50% de que se movam para pólos opostos. Ao final da meiose,
quando o número de cromossomos é finalmente reduzido, metade dos gametas deverá conter uma combinação
parental de alelos (A B ou a b) e metade deverá conter uma nova combinação (A b ou a B). Juntos, existirão
quatro tipos de gametas, cada um sendo um quarto do total. Esta igualdade de frequências gaméticas é um
resultado do comportamento independente dos dois pares de cromossomos durante a meiose I. O princípio
da distribuição independente, de Mendel, refere-se à separação anafásica de genes em pares diferentes de
cromossomos. Mais adiante veremos que os genes no mesmo par de cromossomos não se distribuem
independentemente. Em vez disso, como eles estão fisicamente ligados uns aos outros, tendem a ir juntos através
da meiose, violando o princípio da distribuição independente.
Pontos Importantes: Os genes estão situados em cromossomos. A disjunção dos cromossomos
durante a meiose é responsável pela segregação e distribuição independente dos genes.
41
GENES LIGAODOS AO SEXO EM SERES HUMANOS
O desenvolvimento da teoria cromossômica dependeu da descoberta da mutação para olho branco
na Drosophila. A análise subsequente demonstrou que esta mutação era um alelo recessivo de um gene ligado
ao X. Embora alguns de nós creditem este episódio importante na história da genética a uma extraordinária
sorte, a descoberta de Morgan da mutação para olhos brancos não foi assim tão marcante. Tais mutações
estão entre as mais fáceis de se detectar porque se apresentam imediatamente em machos hemizigotos. Ao
contrário, as mutações autossômicas recessivas só se apresentam quando dois alelos mutantes se juntam em
um homozigoto, um evento muito mais improvável.
Nos seres humanos, também, as características recessivas ligadas ao X são muito mais facilmente
identificadas que as características autossômicas recessivas. Um homem precisa herdar apenas um alelo
recessivo para apresentar uma característica ligada ao X. Entretanto, uma mulher precisa herdar dois, um de
cada genitor. Assim, a preponderância de pessoas que apresentam características ligadas ao X é masculina.
Hemofilia, um Distúrbio de Coagulação Sanguínea Ligado ao X
Nos seres humanos, um determinado tipo de hemofilia é um dos exemplos mais bem conhecidos de
uma característica ligada ao X. As pessoas com esta doença são incapazes de produzir um fator necessário
para a coagulação do sangue. Os cortes e feridas nos hemofílicos continuam a sangrar, e se não forem para
dos por tratamento terapêutico, podem causar a morte. Quase todos os indivíduos afetados por hemofilia
ligada ao X são homens. Outros distúrbios de coagulação sanguínea são encontrados tanto em homens quanto
em mulheres porque são devidos a mutações em genes autossômicos.
O caso mais famoso de hemofilia ligada ao X ocorreu na família imperial russa no início do
século XX (Fig.21). O czar Nicolau e a czarina Alexandra tiveram quatro filhas e um filho. O filho,
Alexis, sofria de hemofilia. A mutação ligada ao X responsável pela doença de Alexis foi transmitida
por sua mãe, que era uma heterozigota para o gene. A czarina Alexandra era neta da rainha Vitória da GrãBretanha, que também era portadora. Os registros de heredograma mostram que Vitória transmitiu o alelo
mutante para três de seus nove filhos: Alice, que era mãe de Alexandra, Beatrice, que teve dois filhos com
a doença, e Leopoldo, que tinha a doença. O alelo que Vitória portava evidentemente surgiu como uma
mutação nova nas células germinativas de um de seus genitores ou em um ancestral materno mais distante.
Alguns acreditam que esta mutação mudou o curso da história, contribuindo para a queda da dinastia
Romanov na Rússia e o subsequente surgimento do estado soviético.
Daltonismo, um Distúrbio Visual Ligado ao X
42
Nos seres humanos, a percepção a cores é mediada por proteínas que absorvem a luz nas células
especializadas dos cones da retina no olho. Três destas proteínas foram identificadas, uma para absorver a
luz azul, uma para absorver a luz verde, e uma para absorver a luz vermelha. O daltonismo pode ser
causado por uma anomalia em qualquer uma destas proteínas receptoras. O tipo clássico de daltonismo,
envolvendo a má percepção da luz vermelha e da verde, segue um padrão de herança ligada ao X. Cerca de
5 a 10 por cento dos homens têm daltonismo verde-vermelho; entretanto, uma fração muito menor de mulheres, menos de l por cento, tem este distúrbio, sugerindo que os alelos mutantes são recessivos. Os
estudos moleculares mostraram que existem dois genes distintos para percepção de cores no cromossomo
X. Um codifica o receptor para a luz verde, e o outro codifica o receptor para a luz vermelha. A análise
detalhada demonstrou que estes dois receptores são estruturalmente muito semelhantes, provavelmente
devido aos genes que os codificam terem evoluído de um gene ancestral de recepção de cores. Um
terceiro gene para a percepção de cores, o que codifica o receptor para a luz azul, está situado em um
autossomo.
Fig. 21 (a) A família imperial russa do czar
Nicolau II. (b) Hemofilia ligada ao X nas
famílias reais da Europa. Através de
casamentos, o alelo mutante para hemofila
foi transmitido da família real inglesa para
as famílias reais alemã, russa e espanhola.
Na Fig. 22, o daltonismo é usado para ilustrar os procedimentos para calcular o risco de herdar
uma condição recessiva ligada ao X. Uma heterozigota (portadora), como a III-4 na figura, tem uma
chance de 1/2 de transmitir o alelo mu-tante para sua prole. Entretanto, o risco de que uma determinada
criança seja daltônica é de apenas 1/4, pois a criança tem que ser do sexo masculino para manifestar a
característica. A mulher IV-2 no heredograma pode ser portadora do alelo mutante para daltonismo,
43
pois sua mãe era. Esta incerteza sobre o genótipo de IV-2 introduz um outro fator de 1/2 no risco de
ter um filho daltônico. Logo, o risco para seu filho é de 1/4 X 1/2 = 1/8.
Fig. 22 Análise de um
heredograma
mostrando
a
segregação do daltonismo ligado
ao X.
A Síndrome do X Frágil e Retardo Mental
Nos seres humanos, muitos casos de retardo mental parecem seguir um padrão ligado ao X de
herança. A maioria deles está associada a uma anomalia citológica que é detectável nas células que foram
cultivadas na ausência de alguns nucleotídeos, os blocos estruturais do DNA. Esta anomalia, uma
constrição perto da ponta do braço longo do cromossomo X, dá a impressão de que a ponta está prestes a
se destacar do resto do cromossomo (Fig. 23), daí o nome cromossomo X frágil. As características
clínicas da síndrome do X frágil variam consideravelmente, dificultando o diagnóstico. A maioria dos
pacientes apresenta uma significativa deficiência mental, e alguns exibem anomalias faciais e
comportamentais. Tanto homens quanto mulheres podem ser afetados. Entre a prole, a incidência de
síndrome de X frágil é de cerca de um em 2.000.
A síndrome do X frágil foi descrita como um distúrbio dominante ligado ao X com penetrância
incompleta. As mulheres afe-tadas são heterozigotas para o cromossomo X frágil, e os homens afetados são
hemizogotos para esta síndrome. Entretanto, alguns portadores do cromossomo, tanto homens quanto
mulheres, são assintomáticos para o distúrbio (Fig. 23). Esta falta de penetrância completa complica a
análise de heredogramas e dificulta a consulta genética.
Foram usadas técnicas moleculares para isolar e analisar o sítio X frágil. Ele consiste em uma
curta unidade repetida de DNA adjacente ao FMR1 (Petardo Mental de X Frágil), um gene de função
desconhecida. Esta unidade repetida varia de comprimento, sendo muito maior nos indivíduos que
44
apresentam o distúrbio. Uma combinação de análises genéticas e moleculares demonstrou que a unidade
repetida tende a aumentar de tamanho, provavelmente como resultado de um defeito de replicação
cromossômica. Esta instabilidade explica por que as pessoas que não apresentam o distúrbio podem ter
filhos que o apresentam. A unidade repetida pode se expandir na linhagem germinativa da mãe e ser
transmitida para os filhos, que a terão em todas as suas células somáticas. Embora os detalhes fisiológicos
não sejam conhecidos, uma unidade expandida parece estar associada à modificação química do DNA
vizinho. Esta modificação tem um efeito adverso na expressão do gene FMR1, e é provavelmente a
causa da sín-drome do X frágil.
Genes no Cromossomo Y Humano
Fig. 23. O cromossomo X frágil, (a) Uma mulher
(esquerda) mostrando o X frágil e um cromossomo X
normal, e um homem (direita) mostrando o X frágil e
um cromossomo Y normal, (b ) Um heredograma
mostrando a herança da síndrome do X frágil. O homem
assintomático II-1 é portador, indicando que a condição
tem penetrância incompleta, (c) A base molecular da
síndrome do X frágil. A mutação no cromossomo X
frágil é devida a uma expansão de uma região de
repetições no DNA flanqueador do gene FMR1. A
modificação química do DNA ao redor destas
repetições afeta adversamente a expressão do gene
FMR1.
Poucos genes foram localizados no cromossomo Y humano. Esta falha em encontrar genes ligados ao
Y é um tanto surpreendente, pois uma mutação em um destes genes deve ter um efeito fenotípico
imediato no homem que o possui. Além disso, tal mutação deve ser passada para todos os filhos deste
homem, mas para nenhuma de suas filhas. Um gene ligado ao Y deve, portanto, ser o tipo de gene mais fácil de
identificar em um heredogra-ma convencional. Hoje em dia, entretanto, apenas alguns genes ligados ao Y
foram descobertos. Um é reponsável pela síntese de uma substância que especifica a masculinidade,
chamada antígeno H-Y, encontrado nas superfícies celulares. Um outro está envolvido na produção de um
fator que é crítico para a diferenciação dos testículos e a subsequente aquisição de características sexuais
45
masculinas. Os avanços na genética molecular forneceram novas técnicas para identificar outros genes
ligados ao Y, mas, mesmo com elas, a opinião de que o cromossomo Y tem menos genes que qualquer outro
cromossomo humano não mudará.
Genes nos Cromossomos X e Y
Alguns genes estão presentes tanto no cromossomo X quanto no Y, a maioria próxima às pontas dos
braços curtos. Os alelos destes genes não seguem um padrão de herança distinto de ligado ao X ou ao Y. Em
vez disso, eles são transmitidos de mães e pais para filhos e filhas igualmente, imitando a herança de um gene
autossômico. Tais genes são, portanto, chamados de genes pseudo-autossômicos. Nos homens, as regiões
que contêm estes genes parecem mediar o parcamente entre os cromossomos X e Y.
Pontos Importantes: Os distúrbios causados por mutações recessivas ligadas ao X, tais como
hemofilia e daltonismo, são mais comuns nos homens que nas mulheres. Nos humanos, o
cromossomo Y porta poucos genes. Alguns destes genes também são portados pelo cromossomo X.
CROMOSSOMOS SEXUAIS E DETERMINAÇÃO DO SEXO
No reino animal, o sexo é talvez o fenótipo mais conspícuo. Os animais com machos e fêmeas
distintos são sexualmente dimórficos. Às vezes este dimorfismo é estabelecido por fatores ambientais. Em
uma espécie de tartarugas, por exemplo, o sexo é determinado pela temperatura. Os ovos que foram
incubados acima de 30°C deram fêmeas, enquanto os que foram incubados em temperatura mais baixa
deram machos. Em muitas outras espécies, o dimorfismo sexual é estabelecido por fatores genéticos, em
geral envolvendo um par de cromossomos sexuais.
DETERMINAÇÃO DO SEXO EM SERES HUMANOS
A descoberta de que as mulheres são XX e os homens XY sugere que o sexo possa ser
determinado pelo número de cromossomos X ou pela presença ou ausência de um cromossomo Y.
Como sabemos, a segunda hipótese é a correia. Em humanos e em outros mamíferos placentários, a
masculinidade é devida a um efeito dominante do cromossomo Y (Fig. 24). A evidência deste fato vem do
estudo de indivíduos com um número anormal de cromossomos sexuais. Os seres XO se desenvolvem
como mulheres, e os XXY se desenvolvem como homens. O efeito dominante do cromossomo Y é
manifesto no início do desenvolvimento, quando direciona as gônadas primordiais para se
desenvolverem em testículos. Uma vez formados os testículos, eles secretam testosterona, um hormô-nio
que estimula o desenvolvimento das características sexuais secundárias.
46
Pesquisas recentes localizaram o fator determinante de testículos (TDF — Testís-determining
Factor) no cromossomo Y humano. Este fator parece corresponder a um gene chamado SRY(Sexdetermining Region Y— região do Y determinante do sexo), situado fora da região pseudo-autossômica no
braço curto do cromossomo. A descoberta do SRY foi possível pela identificação de indivíduos incomuns
cujo sexo era inconsistente com sua constituição cromossômica — XX homens e XY mulheres (Fig. 24).
Alguns dos homens XX foram encontrados levando um pequeno trecho do cromossomo Y inserido em um
dos cromossomos X. Este trecho evidentemente leva um gene responsável pela masculinização. Algumas
mulheres XY foram encontradas possuindo um cromossomo Y incompleto. A parte do cromossomo Y
que estava faltando correspondeu ao trecho que estava presente nos homens XX. Sua ausência nas
mulheres XY aparentemente impediu que desenvolvessem testículos. Estas linhas complementares de
evidência mostraram que um segmento particular do cromossomo Y era necessário para o desenvolvimento
masculino. Análises moleculares subsequentemente identificaram o gene SRY neste segmento determinante
da masculinização. Pesquisas adicionais mostraram que um gene SRY está presente no cromossomo Y do
camundongo, e que — como o gene SRY humano — especifica um desenvolvimento masculino.
Após terem se formado os testículos, a secreção de testosterona inicia o desenvolvimento das
características sexuais masculinas. A testosterona é um hormônio que se liga aos receptores de muitos
tipos de células. Uma vez ligado, o complexo hormônio-receptor transmite um sinal para a célula,
instruindo-a a como se diferenciar. A diferenciação de muitos tipos de células leva ao
desenvolvimento de características distintamente masculinas, tais como musculatura forte, barba e voz
grossa. Se o sistema de sinalização de testosterona falha, estas características não aparecem, e o
indivíduo se desenvolve como mulher. Um motivo da falha é a inabilidade em fazer o receptor de
testosterona (Fig. 25). Os indivíduos XY com esta deficiência bioquímica inicialmente se desenvolvem
como homens, os testículos são formados e a testosterona é produzida. Entretanto, a testosterona não tem
efeito porque não pode atingir suas células-alvo para transmitir o sinal desenvolvimental. As pessoas que
não têm receptor de testosterona mudam de sexo durante o desenvolvimento em-briológico e adquirem
características sexuais femininas. Entretanto, elas não desenvolvem ovários e são portanto estéreis. Esta
síndrome, chamada feminização testicular, resulta de uma mutação em um gene ligado ao X, tf m, que
codifica o receptor de testosterona. A mutação tfm é transmitida pelas mães para os filhos (que são
fenotipicamente mulheres) em um padrão tipicamente ligado ao X.
47
Determinação do Sexo na Drosophila
Fig. 24 O processo de determinação do
sexo
em
seres
humanos.
O
desenvolvimento sexual masculino na
produção do fator determinante
testicular (TDF) por um gene no
cromossomo Y. Na ausência deste fator,
o embrião se desenvolve como uma
mulher.
O cromossomo Y na Drosophila, diferentemente do que ocorre em humanos, não tem papel na
determinação do sexo. O sexo da mosca é determinado pela proporção de cromossomos X em relação
aos autossomos. Este mecanismo foi primeiro demonstrado por Bridges em 1921 por meio de uma
análise das moscas com constituições cromossômicas incomuns.
Fig. 25 Evidência localizando o gene para o fator
determinante testicular (TDF) no braço curto do
cromossomo Y em homens normais. O TDF é o
produto do gene SRY. Nos homens XX, uma
pequena região contendo este gene foi inserida em
um dos cromossomos X, e nas mulheres XY, ela
foi suprimida do cromossomo Y.
As moscas diplóides normais têm um par de cromossomos sexuais, XX ou XY, e três pares de
autossomos, em geral indicados como AA; aqui, cada A representa um conjunto ha-plóide de
autossomos. Por artifícios genéticos, Bridges obteve moscas com números anormais de cromossomos
(Quadro 4). Ele observou que sempre que a proporção de X para A era 1,0 ou maior, a mosca era fêmea,
48
e sempre que era 0,5 ou menor, a mosca era macho. As moscas com uma proporção X:A entre 0,5 e 1,0
desenvolveram características de ambos os sexos, e Bridges as chamou de intersexos. Em nenhuma
destas moscas o cromossomo Y teve algum efeito no fenóti-po sexual. Entretanto, ele era necessário
para a fertilidade masculina.
Sabemos hoje que um gene ligado ao X chamado Sexo-le-tal (Sxl) tem um papel importante no
sistema de determinação do sexo da Drosophila (Fig. .15). Uma rede elaborada de outros genes ligados
ao X, trabalhando com fatores já presentes nos ovócitos de Drosophila, estabelecem o nível de
atividade de Sxl em um zigoto. Se a proporção X: A for maior ou igual a um, o gene Sxl é ativado e o
zigoto se desenvolve como fêmea. Se for menor ou igual a 0,5, o gene Sxl é inati-vado e o zigoto se
desenvolve como macho. Uma proporção entre 0,5 e 1,0 causa uma mistura de sinais, e o zigoto se
desenvolve com características tanto masculinas quanto femininas. Este sistema tem a incrível
habilidade de contar cromossomos, computar a proporção de X para A, e então ligar ou desligar o
interruptor Sxl. A expressão imprópria de Sxl — desligado em fêmeas ou em machos — leva à morte
embrionária, donde o nome do gene, letal sexual.
Fig. 26 Feminização testicular, uma condição causada por uma mutação ligada ao X, tfm, que
impede a produção do receptor de testosterona.
DETERMINAÇÃO DO SEXO EM OUTROS ANIMAIS
Em Drosophila e em seres humanos, os machos produzem dois tipos de gametas, os portadores de X
e os de Y. Por este motivo, eles são chamados de sexo heterogamético. Nestas espécies, as fêmeas são o
sexo homogamético. Nas aves, nas borboletas, e em alguns répteis, esta situação é revertida (Fig. 27). Os
machos são homogaméticos (em geral indicados por ZZ) e as fêmeas são heterogaméticas (ZW). Entretanto,
pouco se sabe sobre o mecanismo de determinação do sexo no sistema de cromossomos sexuais Z-W.
49
Quadro 4. Proporção de Cromossomos X para Autossomos e o Fenótipo Correspondente em
Drosophila
Nas abelhas, o sexo é determinado pelo fato de o animal ser haplóide ou diplóide (Fig. 27). Os
embriões diplóides, que se desenvolvem de ovócitos fertilizados, tornam-se fêmeas. Os embriões
haplóides, que se desenvolvem de ovócitos não fertilizados, tornam-se machos. O fato de uma determinada fêmea se desenvolver ou não em uma forma reprodutiva (rainha) depende de como ela foi
alimentada quando larva. Neste sistema, uma rainha pode controlar a proporção de machos e fêmeas
regulando a proporção de ovócitos não-fertilizados que ela põe. Como este número é pequeno, a maioria
da prole é fêmea, embora estéril, e serve como operárias na colmeia. Em um sistema haplo-diplóide de
determinação sexual, os ovócitos são produzidos por meiose na rainha e os espermatozóides são
produzidos por mitose no macho. Este sistema garante que os ovócitos fertilizados terão o número
cromossômico diplóide e os não-fertilizados terão o número haplóide.
Fig. 27 A alternância de Sxl na
determinação do sexo na Drosophila. O
gene Sxl é ligado nos zigotos nos quais a
proporção X:A é 1,0 e desligado em zigotos
nos quais a proporção X:A é 0,5. A
expressão do gene Sxl faz com que o zigoto
se desenvolva em uma fêmea, enquanto a
não expressão deste gene faz com que o
zigoto se desenvolva em um macho.
50
Fig. 28 Determinação do sexo em aves. A
fêmea é heterogamética (ZW) e o macho é
homogamético (ZZ). O sexo da prole é
determinado por qual cromossomo
sexual, Z ou W, é transmitido para a
fêmea.
Fig. 29 A determinação do sexo nas
abelhas. As fêmeas, que são derivadas
de ovócitos fertilizados, são diplóides, e
os machos, que são derivados de
ovócitos não-fertilizados, são haplóides.
Algumas vespas também têm o método haplo-diplóide de determinação do sexo. Nestas espécies, os machos
diplóides são às vezes produzidos, mas eles são sempre estéreis. A análise genética detalhada em uma espécie,
Bracon hebetor, indicou que os machos diplóides são homozigotos para um locus determinante do sexo, chamado
X; as fêmeas diplóides são sempre heterozigotas para este locus. Evidentemente, o locus do sexo em Bracon tem
muitos alelos. Os cruzamentos entre machos e fêmeas não aparentados, portanto, quase sempre produzem fêmeas
diplóides heterozigotas. Entretanto, quando os machos são aparentados, há uma apreciável chance de que sua prole
seja homozigota para o locus sexual, e neste caso se desenvolvem em machos estéreis.
COMPENSAÇÃO DE DOSE DE GENES LIGADOS AO X
O desenvolvimento animal geralmente é sensível a um desequilíbrio no número de genes.
Normalmente, cada gene está presente em duas cópias. Os desvios desta condição, seja para cima ou para
baixo, podem causar fenótipos anormais e às vezes até mesmo a morte. É portanto uma incógnita que
tantas espécies devam ter um sistema de determinação do sexo baseado em fêmeas com dois
cromossomos X e os machos com apenas um. Nestas espécies, como é acomodada a diferença numérica
de genes ligados ao X? A priori, dois mecanismos podem compensar esta diferença: (1) cada gene liga-I
do ao X funciona duas vezes mais em machos que em fême-1 as, ou (2) uma cópia de cada gene ligado
51
ao X é inativada nas l fêmeas. Várias pesquisas mostraram que ambos os mecanismos são usados, o
primeiro em Drosophila e o segundo em mamíferos.
HIPERATIVAÇÃO DE GENES LIGADOS AO X EM MACHOS DE DROSOPHILA
Na Drosophila, a compensação de dose de genes ligados ao X é obtida por um aumento na
atividade destes genes nos machos. Este fenómeno, chamado hiperativação, envolve o gene letal
sexual, que também tem um papel importante na determinação do sexo (Fig. 30). O gene Sxl é ligado
nas fêmeas e desligado nos machos. Quando o produto gênico Sxl está ausente (como nos machos), um
complexo de proteínas diferentes se liga a muitos sítios no cromossomo X e ativa uma duplicação da
atividade gênica. Quando o produto do gene Sxl está presente (como nas fêmeas), este complexo
proteico não se liga, e a hiperativação de genes ligados ao X não ocorre. Deste modo, a atividade
total de genes ligados ao X nos machos e fêmeas é aproximadamente igualada.
INATIVAÇÃO DE GENES LIGADOS AO X EM FÊMEAS DE MAMÍFEROS
Nos mamíferos placentários, a compensação de dose de genes ligados ao X é obtida pela
inativação de um dos cromossomos X das fêmeas (Fig. 31). Este mecanismo foi inicialmente proposto em
1961 pela geneticista britânica Mary Lyon, que o deduziu de estudos em camundongos. As pesquisas subsequentes de Lyon e outros mostraram que o evento de inativação ocorre quando o embrião de
camundongo consiste em alguns milhares de células. Nesta época, cada célula toma uma decisão
independente para silenciar um de seus cromossomos X. O cromossomo a ser inativado é escolhido ao acaso. Uma
vez escolhido, entretanto, ele permanece inativo em todos os descendentes desta célula. Assim, as fêmeas de mamíferos são mosaicos genéticos contendo dois tipos de clo-nes celulares. O cromossomo X herdado maternamente
é inativado em mais ou menos metade destes clones, e o paterna-mente herdado é inativado na outra metade. Uma
fêmea que é heterozigota para um alelo ligado ao X é portanto capaz de apresentar dois fenótipos diferentes.
Fig.30 Compensação de dose em
Drosophila. O único cromossomo X
nos machos é hiperativado devido a
um complexo proteico que se liga ao
cromossomo. Nas fêmeas, onde o
gene Sxl é expresso, este complexo
não se liga aos cromossomos, e estes
cromossomos não são hiperativados.
52
Um dos melhores exemplos deste mosaicismo fenotípico vem do estudo da coloração do pêlo em
gatos e camundongos. Em ambas as espécies, o cromossomo X porta um gene para pigmentação do pêlo.
As fêmeas heterozigotas para alelos diferentes deste gene mostram placas de pêlos claros e escuros. As
placas claras expressam um alelo; as escuras expressam o outro. Em gatos, onde um alelo produz o
pigmento preto e o outro produz pigmento laranja, este fenótipo em placas é chamado de tortoiseshell. Cada
placa de pêlos define um clone de células produtoras de pigmento, ou melanócitos, que foram derivadas
por mitose de uma célula precursora presente no momento da inativação do cromossomo X.
Muitos aspectos dos mecanismos de inativação do cromossomo X ainda são um mistério. A análise
genética mostrou que tanto em humanos quanto em camundongos ela começa em um ponto do braço longo
do cromossomo X e se espalha em ambos os sentidos para as pontas dos cromossomos . O local iniciador é
chamado centro de inativação do X (XIC).
Um cromossomo X que foi inativado não se parece com os outros cromossomos nem atua como
eles. As análises químicas mostram que seu DNA é modificado pela adição de numerosos grupos metila.
Além disso, ele se condensa em uma estrutura fortemente corada chamada corpúsculo de Barr, em
homenagem ao geneticista canadense Murray Barr, que primeiro o observou. Esta estrutura fica ligada à face
interna da membrana nuclear, onde se replica em momento diferente do dos outros cromossomos da
célula. O cromossomo X inativado permanece neste estado alterado em todos os tecidos somáticos.
Entretanto, nos tecidos germinativos ele é reativado, talvez porque duas cópias de alguns genes ligados ao X
são necessárias para o término bem-sucedido da ovocitogênese.
Estudos citológicos identificaram seres humanos com mais de dois cromossomos X. Para a maior
parte, estas pessoas são fenotipicamente mulheres normais, aparentemente porque todos os seus
cromossomos X, menos um, estão inativados. Em geral, todos os X inativados se unem em um só corpúsculo
de Barr. Estas observações sugerem que as células podem ter uma quantidade limitada de algum fator
necessário para evitar a inativação do X. Uma vez que este fator tenha sido usado para manter um
cromossomo X ativo, todos os outros entram no processo de inativação.
53
Pontos Importantes: Na Drosophila, a compensação de dose para genes ligados ao X é obtida
pela hiperatividade do único cromossomo X nos machos. Em mamíferos, a compensação de dose
é obtida inativando um dos dois cromossomos X em mulheres.
Fig. 30 Inativação do cromossomo X em
mamíferos. Um dos cromossomos X nas fêmeas
XX é inativado em cada célula do embrião inicial.
Na linhagem germinativa, os cromossomos X
inativados são subsequentemente reativados
durante a ovocitogênese.
54
GENÉTICA EM MEDICINA
A Natureza em nenhuma outra parte está mais acostumada a apresentar seus mistérios secretos que
nos casos onde ela apresenta traços de seus trabalhos fora do que é normal; e nem há um modo melhor para
avançar na prática adequada da medicina do que entregar nossas mentes à descoberta da lei usual da
Natureza pela cuidadosa investigação dos casos deformas raras. Foi encontrado, em quase todas as coisas,
que o que elas contêm de útil ou de natureza aplicável dificilmente é percebido, a menos que estejamos
privados delas, ou elas estejam de algum modo perturbadas.
Carta escrita por William Harvey em 1657 (citada por A. Garrod em 1928 em um artigo sobre
doenças raras. Lancet, i, pp. 1055-1060, 1928)
A genética moderna teve um profundo impacto na medicina. Embora as conexões entre algumas
doenças e a herança tivessem sido feitas há séculos, uma das primeiras e mais importantes ligações entre os
novos princípios mendelianos descobertos e as doenças foi publicada por Sir Archibald Garrod em 1902.
Este médico inglês estava estudando doenças metabólicas raras que se transmitem em famílias, e
assim pareciam ser herdadas. Um destes distúrbios foi a alcaptonúria, um distúrbio metabólico no qual uma
substância chamada alcaptona se acumula nas células e tecidos e é excretada na urina. A alcaptona,
apropriadamente conhecida como ácido homogentísico, é um produto metabólico intermediário formado
pelo metabolismo do aminoácido fenilalanina. Normalmente, o ácido homogentísico é convertido em outro
composto (maleilacetoacetato), mas neste caso a enzima que possibilita a conversão está ausente. O
ácido homogentísico então se acumula e é oxidado em um produto escuro que se acumula em áreas do
corpo ricas em cartilagem, tornando escuros as orelhas, articulações e outros tecidos. A urina também fica
escura quando exposta ao oxigénio. Embora este acúmulo de ácido homogentísico seja uma condição
clínica relativamente benigna, causando no máximo uma moderada artrite, ele persiste durante a vida do
paciente.
Garrod, após discussões com William Bateson, um dos pioneiros das ideias mendelianas, concluiu
que a alcaptonúria era herdada de acordo com princípios mendelianos, e que fatores mendelianos
55
controlavam a química celular. Garrod foi o primeiro a associar um gene mutante defeituoso a um bloqueio em
uma via metabólica causando um distúrbio herdado. Em reconhecimento por suas contribuições, Garrod é
geralmente reconhecido como o "pai da genética bioquímica".
A brilhante análise de Garrod sobre a alcaptonúria hoje é considerada a base da genética médica
moderna. Embora muitos distúrbios hereditários sejam raros, sua compreensão nos possibilita um insight
de como os genes controlam os processos celulares normais. Por exemplo, os estudos da alcaptonúria, onde
um processo metabólico normal é perturbado, levou a descobertas importantes de como o aminoácido
fenilalanina é normalmente metabolizado. Os indivíduos com um distúrbio hereditário chamado
fenilcetonúria, ou simplesmente PKU, são incapazes de converter normalmente a fenilalanina em outro
aminoácido, a tirosina, devido à falta da enzima necessária para esta reação. Como resultado, os derivados da
fenilalanina que são tóxicos para o sistema nervoso central se acumulam na corrente sanguínea e causam
grave retardo mental. Entretanto, um cientista determinou a base molecular do retardo mental em indivíduos
com PKU, e um tratamento eficaz logo foi estabelecido. Quando a PKU é diagnosticada ao nascimento, a
criança é colocada em uma dieta com níveis controlados de fenilalanina — suficientemente alta para apoiar
o crescimento normal mas baixa o suficiente para evitar a toxidez das células nervosas.
Desde a época de Garrod, os pesquisadores fizeram grandes esforços para estabelecer ligações
entre genes defeituosos e doenças. Discutiremos algumas das principais ligações aqui e voltaremos a
muitas delas durante o texto. Os genes nas pessoas normais são chamados de genes tipo selvagem; aqueles
com defeitos que resultam na síntese de produtos não-funcionais são chamados de genes mutantes. Eles
resultam em condições anormais ou mutantes. Os defeitos em genes mutantes são chamados de mutações.
A hemofilia, ou doença do sangramento, foi um dos primeiros distúrbios humanos a serem ligados a
um gene mutante e ao seu produto não-funcional. No passado, os indivíduos com este defeito herdado de
coagulação sanguínea em geral morriam durante a infância. Hoje em dia, eles em geral vivem uma vida
normal graças a tratamentos eficazes com fator de coagulação produzido por culturas de células de
mamíferos geneticamente modificadas .
O gene mutante que é responsável pela doença de Hunting-ton, um distúrbio neurológico fatal, foi
isolado em 1993 após intensa pesquisa de 10 anos. O gene mutante responsável pela fibrose cística foi
também isolado e seu produto gênico identificado. Com a compreensão dos mecanismos de doença veio a
oportunidade de tratamento.
A terapia gênica — a introdução de genes normais nas células de pacientes com genes defeituosos —
oferece grandes promessas como tratamento eficaz de doenças herdadas. Infelizmente, hoje em dia a terapia
gênica ainda não concretizou essas promessas. A terapia gênica tem sido usada em combinação com outros
56
tratamentos em crianças com um distúrbio devastador do sistema imune chamado doença grave de
imunodeficiência combinada, mas os resultados têm sido desestimulantes. Os genes introduzidos foram
expressos por apenas um curto período de tempo. As tentativas clínicas de terapia gênica estão atualmente
sendo feitas em pacientes com vários tipos de câncer, doença de Gaucher, hipercolesterolemia familial,
hemofilia, anemia de Fanconi, síndrome de Hunter, e a síndrome da deficiência imune adquirida (AIDS).
Embora existam motivos para se esperar que a terapia gênica seja finalmente bem sucedida no
tratamento destes e de outros distúrbios, os resultados atuais indicam que são necessárias mais pesquisas para
se determinar como e por que os genes são desligados logo após entrarem em novas células hospedeiras.
Algumas formas de câncer são familiais (elas ocorrem frequentemente dentro de famílias) ou
hereditárias; outras são não hereditárias e ocorrem esporadicamente entre todos os membros de uma
população. Entretanto, todos os cânceres são doenças genéticas no sentido de que são causadas por
mudanças na informação genética que é transmitida para as células filhas. A evidência disponível indica
que todos os cânceres resultam do acúmulo de danos aos genes que controlam ou influenciam a
multiplicação celular, a diferenciação celular e processos correlatos. Embora existam centenas de tipos
diferentes de câncer, todos têm uma coisa em comum, a perda do controle normal da multiplicação celular.
Os genes mutantes que causam um risco aumentado de câncer estão sendo identificados e estudados
intensamente. Um destes genes, chamado BRCA1 para gene de câncer de mama l, resulta em aumento de
suscetibilidade ao câncer de mama e ovário em algumas famílias. O gene BRCA1 foi isolado, caracterizado e
mapeado no cromossomo 17. Um segundo gene de câncer de mama, o BRCA2, foi subsequentemente
identificado. Eles se juntam a uma longa lista de genes que causam câncer familial, incluindo o tumor de
Wilms, um câncer renal em crianças, e a síndrome de Gardner, um tipo de câncer do cólon. À medida que
aprendemos mais sobre o funcionamento destes genes de câncer em situações normais e anormais, chegamos
mais perto de tratamentos terapêuticos eficazes.
Entretanto, como no caso de outros genes identificados por esta estratégia, é um longo caminho
entre isolar o gene, identificar seu produto e determinar o que o produto faz na célula. Mesmo com a
informação sobre o funcionamento de um gene, criar uma terapia é outro problema de grandes proporções.
Por exemplo, o gene para doença de Huntington foi isolado em 1993, e seu produto proteico, a huntingtina,
foi identificado logo após. Mas, nos cinco anos seguintes, a função da huntingtina ainda era desconhecida.
Nancy Wexler, que trabalhou na descoberta da doença de Huntington, compartilha suas ideias conosco
sobre os aspectos científico, pessoal e social da doença. Uma vez que a função do gene da doença de
Huntington seja totalmente compreendida, ainda poderá haver um longo tempo até que uma terapia efetiva
para esta doença devastadora seja estabelecida.
57
Em contraste com a doença de Huntington, sabemos bastante sobre a função do produto do gene da
fibrose cística, chamado CFTR — regulador da condutância transmembranar da fibrose cística (em inglês:
cystic yibrosis íransmembrane conductance regulator). Este conhecimento sugere que algum dia seja possível tratar a fibrose cística pela terapia gênica, isto é, introduzindo cópias funcionais do gene CFTR nas células
de indivíduos com versões defeituosas do gene. Em uma estratégia, uma forma normal do gene CFTR é
inserida em um vírus comum do resfriado que tenha sido geneticamente alterado, de modo que ele não possa
completar seu ciclo de vida normal. Este vírus modificado é então aplicado às passagens nasais de
pacientes com CF (fibrose cística). O envoltório que encapsula o vírus da gripe funciona como o veículo
de transferência do gene normal CFTR para as células do epitélio nasal. Uma vez nas células, o gene
CFTR deve dirigir a síntese de um produto gênico normal, e as células, previamente deficientes de CFTR,
devem recuperar parte de seu funcionamento normal. Infelizmente, até agora, este tipo de terapia gênica não
se demonstrou muito eficaz. Muito poucas células na verdade adquirem cópias funcionais do gene, e os níveis
de CFTR produzidos nas células de pacientes com CF tratados são muito baixos para eliminar os sintomas
da doença. Além disso, este tipo de terapia gênica não corrige o defeito pan-creático desta doença nem corrige
os defeitos causados pela CF em outras células. Embora os experimentos iniciais de terapia gênica não
tenham tido sucesso, parece seguro prever que, no futuro — próximo, esperamos —, a terapia gênica
forneça um tratamento eficaz para a fibrose cística.
O isolamento e caracterização dos genes mutantes BRCA1 e BRCA2 que aumentam o risco de
câncer de mama estão criando aspectos sociais e éticos importantes. Por exemplo, BRCA1 é um gene muito
grande, e mais de 100 mutações diferentes dele já foram identificadas em humanos. É uma tarefa cara e
trabalhosa triar mulheres quanto a possíveis variantes deste gene de modo rotineiro. Para complicar ainda
mais as coisas, algumas variantes de BRCA1 são benignas e não predispõem a mulher ao câncer de mama.
Portanto, parece que o teste será feito primariamente em mulheres com uma história familial de câncer
de mama. Mesmo nestes casos, entretanto, há o risco de que os testes laboratoriais produzam resultados
falso-positivos ou falso-negativos. Nos casos onde o teste diagnóstico indica que uma mulher está portando
um gene mutante BRCA1 ou BRCA2 que pode predispô-la a desenvolver câncer de mama, que opções de
tratamento podem ser oferecidas? É sempre benéfico para a mulher dizer-lhe que ela é portadora de uma
mutação que a predispõe a desenvolver câncer de mama? A mastectomia profilática é um tratamento
recomendado para portadoras de genes BRCA mutantes, mas este tratamento pode nem sempre ser bem-sucedido. Além disso, como os genes mutantes BRCA também aumentam o risco de câncer de ovário, o
consultor genético deve sugerir que a mulher com mutações BRCA tenha seus ovários removidos? Muitos
58
problemas atormentantes estão associados à identificação e isolamento de genes de câncer de mama, problemas que têm que ser abordados.
Recentemente, os cientistas combinaram técnicas genéticas com aplicações computadorizadas
para desenvolver novas e poderosas tecnologias de chips gênicos que podem ser usados para diagnosticar
rapidamente doenças herdadas, presença de vírus ou mudanças nos padrões de expressão gênica em tecidos
de pacientes com várias doenças. Os chips gênicos podem ser usados para comparar rapidamente os níveis
de expressão de 10.000 ou mais genes em tecidos selecionados de pessoas com uma determinada doença e
de pessoas normais. Os primeiros chips gênicos a se tornarem disponíveis comercialmente foram destinados a
detectar HIV (o vírus que causa a AIDS), para detectar alterações na estrutura do gene humano p53 supressor
tumoral (alterado em vários tipos de câncer) e para quantificar os RNAs produzidos por 7.700 genes
humanos. Outros chips gênicos logo estarão disponíveis. As tecnologias de chips gênicos prometem ampliar
nossa compreensão das relações entre a expressão gênica alterada e as doenças humanas. Este conhecimento
deve ajudar os cientistas a desenvolver métodos eficazes para aliviar a dor e o sofrimento associados a estas
doenças.
A genética teve e continua a ter um profundo impacto no campo da medicina. Os genes defeituosos
colocam os humanos em risco aumentado de desenvolver muitas doenças. O tratamento de distúrbios
herdados usando-se a terapia gênica tem um grande potencial, mas ainda não se demonstrou eficaz.
GENÉTICA E AGRICULTURA MODERNA
Além de seu impacto na medicina, a genética moderna teve um tremendo impacto na agricultura.
Uma das grandes conquistas na agricultura moderna foi a aplicação dos princípios mendelia-nos ao
desenvolvimento do milho híbrido. O milho híbrido é produzido pelo cruzamento de linhagens
endogâmicas diferentes, cada uma das quais com propriedades valiosas, como resistência a doenças, alto teor
de proteínas e açúcar e tolerância à estiagem. Durante o período de 1940 a 1980, a produção média de milho
aumentou em 250%, em grande parte devido ao desenvolvimento e introdução de variedades de milho
híbrido. Nos Estados Unidos, a hibridização resultou em acentuados aumentos de produção em quase todas
as colheitas de alimentos importantes, como cevada, feijão, aveia, arroz e trigo, embora não tão grandes
quanto de milho híbrido.
Dos anos 1950 aos 1970, Norman Borlaug e sua equipe de pesquisadores usaram os princípios da
genética clássica para desenvolver novas linhagens de trigo no México que se desenvolveriam bem em
condições de estresse em geral encontradas em países em desenvolvimento. Seu sucesso em desenvolver este
trigo, bem como outras plantas, lhe deu o Prémio Nobel de 1970. Suas enormes contribuições para a
59
produtividade agricultural, e portanto para a nutrição, em países em desenvolvimento ajudaram a
desencadear a tão divulgada "revolução verde".
O tomate moderno se beneficiou muito da aplicação dos princípios genéticos. Os genes que
conferem resistência a vários patógenos, como fungos e nematódeos, foram introduzidos nas variedades
modernas. A maioria dos genes de resistência veio de similares do tomate que são selvagens. O padrão de
crescimento do tomateiro também foi geneticamente alterado, de modo que a planta é mais arbustiva e
compacta, características que ajudam a manter os frutos fora do chão. Os agricultores desenvolveram uma
grande variedade de tipos morfológicos de tomates. Eles incluem os redondos, medium-sized lemon boys; os
redondos, large beefsteaks; os pequenos, round cherry; em forma de pêra, amarelos e vermelhos; e outros.
Os programas de cruzamentos seletivos produziram galinhas que têm mais carne, crescem mais
depressa, são mais resistentes a doenças e botam mais ovos. Os programas de cruzamentos seletivos
produziram gado, ovelhas e porcos que têm mais carne, crescem mais depressa e são mais eficientes em
converter alimentos em carne, além de melhor adaptados aos ambientes regionais. A produção de leite
pela vaca e seu conteúdo de manteiga também aumentou acentuadamente como consequência dos
cruzamentos seletivos.
Uma das técnicas mais eficazes usadas para melhorar a qualidade das criações é a inseminação
artificial. Um touro conhecido por ser pai de vacas que produzem grandes quantidades de leite ou geram
prole produtora de grande quantidade de carne tem seus espermatozóides coletados a intervalos regulares e
estocado. Este esperma é usado para fertilizar milhares de fêmeas em todo o mundo. Os genes favoráveis
deste touro são assim transmitidos para uma prole numerosa, resultando em grandes aumentos de produção de
leite ou de carne.
Além dos cruzamentos genéticos e seleção de características desejadas que foram usadas por
criadores de plantas e animais durante séculos, as novas tecnologias genéticas já tiveram e continuam a ter um
grande impacto na agricultura. Inserir genes para resistência a insetos ou patógenos em plantios está se
tornando uma importante arma contra organismos que destroem muito do suprimento mundial de alimentos.
A broca do milho é um inseto que ataca o caule do milho, interrompendo o fluxo de nutrientes para as
espigas e diminuindo a produção. Os túneis produzidos pela broca do milho também enfraquecem o
caule, fazendo com que as plantas se quebrem com um vento forte. Em combinação, os efeitos da broca
do milho causam importantes diminuições na produção e despesas para os agricultores de cerca de US$ l
bilhão por ano apenas nos Estados Unidos. No passado, os agricultores tentaram controlar os danos da broca
do milho pulverizando seus campos com inseticidas. Hoje têm outra opção. A bactéria Bacillus
thuringiensis contém um gene codificante de uma proteína que é tóxica para muitos insetos. Subespécies
60
diferentes desta bactéria produzem toxinas que matam insetos diferentes. Uma subespécie produz uma
toxina que mata a broca do milho. O gene para toxina proteica foi isolado da bactéria e inserido nos pés de
milho. Quando a broca se alimenta de plantas portadoras do gene da toxina, ela morre em alguns dias,
enquanto as plantas que não têm o gene ficam desprotegidas.
Uma outra aplicação da genética molecular para a melhoria de plantas e animais é o uso de marcadores
moleculares para determinar as localizações dos genes ou grupos de genes que controlam características
importantes, tais como a taxa de crescimento e a produção. Estas características em geral exibem variação
contínua, por exemplo, da planta de milho mais baixa para a mais alta. Portanto, elas são chamadas de
características quantitativas (QTs), e os genes ou grupos de genes que os controlam são chamados de loci de
características quantitativas. Um locus (plural loci) é simplesmente o sítio ou local de um gene ou de uma
mutação em um cromossomo. Uma vez que os loci de QT tenham sido mapeados em regiões de cromossomos
individuais, os gene-ticistas podem planejar cruzamentos para introduzi-los em variedades importantes de
cultivo ou de criação de animais. Assim, os loci de QT estão sendo amplamente usados para acelerar os
projetos de criação de animais e plantas.
Enquanto a aplicação dos fundamentos da genética ao cultivo de plantas e criação de animais estava
produzindo acentuados aumentos na produtividade agrícola em quase todo o mundo, a produção agrícola na
União Soviética estava estagnada. De 1937 a 1964, a biologia e agricultura na União Soviética eram
dominadas por uma pessoa, Trofim D. Lysenko, um jovem agricultor da Ucrânia. Lysenko acreditava que o
desenvolvimento das plantas e a produtividade agrícola podiam ser amplamente melhorados manipulandose o ambiente. As mudanças induzidas pelo ambiente no crescimento das plantas, segundo Lysenko, seriam
assimiladas pelo material genético e passadas para a geração seguinte. Suas ideias eram semelhantes às
formuladas por Lamark no começo do século XIX. As ideias de Lysenko foram finalmente desacreditadas,
mas só após a produtividade da agricultura na União Soviética ter ficado muito aquém da dos países
desenvolvidos. A era de Lysenko na União Soviética deu uma clara ilustração do perigo de se dar muito
poder a uma única pessoa.
Embora os fundamentos da genética clássica e moderna venham sendo aplicados à agricultura há
menos de 100 anos, não devemos perder de vista o fato de que a espécie humana tem usado a genética na
agricultura por séculos sem o conhecimento formal das leis da herança. Os seres humanos pioneiros fizeram
os primeiros experimentos de "seleção genética" em trigo entre 7.000 a 10.000 anos atrás. As evidências
indicam que quase todos os nossos cultivos atuais de alimentos foram domesticados durante este período
Neolítico inicial, coincidindo com o desenvolvimento dos instrumentos de pedra. Os indivíduos maiores e
mais vigorosos na população foram selecionados como genitores das gerações subsequentes, uma prática que
61
ainda é a principal dos agricultores e criadores de animais. Entretanto, esta prática agora está sendo
suplementada com novos e poderosos instrumentos de genética molecular e engenharia genética. Como resultado, podemos esperar um rápido progresso e grandes conquistas na agricultura no futuro, como foi no
passado.
Pontos Importantes: A genética teve um grande impacto na agricultura. Hoje em dia, tanto o
cruzamento seletivo quanto os novos enfoques de genética molecular estão sendo aplicados ao
desafio de alimentar a população humana mundial em crescimento rápido.
GENÉTICA E SOCIEDADE
As metas do Projeto Genoma Humano, lançadas em 1990, são mapear e seqüenciar toda a
informação genética de humanos e de alguns outros organismos geneticamente importantes por volta de 2005.
O progresso nestas metas já está à frente do cronograma, e uma empresa privada recém-formada anunciou que
seqüenciará todo o genoma humano em apenas três anos, por volta de 2001. Outra empresa privada está
seqüenciando as partes expressas do genoma humano, os genes. As sequências completas dos genomas de 18
bactérias e da levedura Saccharomyces cerevisiae já foram determinadas. A sequência do genoma do verme
Caenorhabditis elegans está 99,9 por cento completa; e as sequências dos genomas da planta Arabidopsis
thaliana e da mosca das frutas Drosophila melanogaster logo estarão disponíveis. O conhecimento da
estrutura de toda a informação genética dos humanos e de outros importantes organismos genéticos terá
profundos efeitos na sociedade. Esta informação terá efeitos acentuados na capacidade dos cientistas de
diagnosticar e criar tratamentos eficazes para doenças humanas. Assim, esta informação deverá ter impacto
muito positivo na saúde humana. Entretanto, também criará complexos problemas morais, éticos e legais
que deverão ser enfrentados pelas pessoas e pela sociedade. Consideremos alguns destes aspectos.
A informação sobre o genoma humano está se acumulando em um ritmo rápido. Em vista destas novas
informações, os cientistas logo poderão ser capazes de fazer previsões importantes sobre a saúde futura das
pessoas: por exemplo, a probabilidade de uma pessoa desenvolver câncer, doença mental ou algum outro
distúrbio com base genética. Um problema complexo que está no cerne desta questão é o acesso às informações
obtidas. A maior parte das pesquisas sobre o genoma humano tem fundos públicos. O público deve ter um acesso
ilimitado à informação? E quanto às seguradoras e patrões? Eles poderão rejeitar um candidato a emprego ou
negar um seguro com base nas informações genéticas. Como a sociedade protegerá a privacidade das pessoas
face à disponibilidade desta nova informação genética?
Um casal jovem tem uma filha com fibrose cística. Para ter esta devastadora doença genética, a criança
herdou duas cópiaspdo gene defeituoso. Logo, cada um dos genitores tinha que ser portador de uma cópia do
gene defeituoso e transmitiu esta cópia para ela. A mãe está novamente grávida, muito embora ela e seu marido
62
saibam que havia um risco de 25% de eles produzirem outra criança afetada. Sua seguradora requisitou um
diagnóstico pré-natal do feto para avaliar sua condição quanto à fibrose cística. Os resultados do teste
mostraram que o feto tinha fibrose cística. Os representantes da seguradora pediram que eles interrompessem a
gestação sob o risco de perderem a cobertura do seguro. O casal se recusou a terminar a gestação, a seguradora
cancelou o seguro e o casal moveu um processo. As seguradoras têm o direito de cancelar as apólices de
seguro ou insistir no término da gestação frente ao nascimento de uma criança com um distúrbio genético de
tratamento caro e geralmente fatal? A corte disse que não. Entretanto, tal questão não termina com uma decisão
legal. De fato, as famílias que correm risco de desenvolver determinadas doenças estão sendo recusadas por
seguradoras.
Entretanto, não é justo dizer que as seguradoras são insensíveis e interessadas apenas no final. Os
distúrbios genéticos tais como fibrose cística são de tratamento caro, geralmente custando centenas de
milhares, ou mesmo milhões de dólares. É apropriado pedir que as seguradoras cubram todo o custo dos
tratamentos? Que direitos ou obrigações as seguradoras têm na prevenção de nascimento dos geneticamente
defeituosos? Que responsabilidades os genitores que estão em risco têm na prevenção de distúrbios genéticos?
À medida que você usar este texto para estabelecer uma fundamentação genética para uma análise
criteriosa, você estará melhor qualificado para avaliá-las.
A genética tem o potencial de aprimorar a qualidade da vida humana. Ao longo deste livro
destacamos como nossos conhecimentos de genética evoluíram e como nossas vidas ficaram melhor
através deste conhecimento. Mas devemos ter em mente alguns infelizes, e mesmo trágicos, usos errados da
genética.
A teoria de Darwin da evolução pela seleção natural diz, entre outras coisas, que os organismos com
características que são benéficas produzem maior número de prole que os organismos com características
menos benéficas. Como resultado, estas características benéficas tornam-se a norma na população. Esta
ideia foi impropriamente aplicada à espécie humana. Francis Galton, primo de Charles Darwin, acreditava
que muitas qualidades físicas e mentais humanas eram herdadas, e portanto sujeitas às forças da seleção. Mas
Galton levou esta ideia um passo adiante. Ele sugeriu que a constituição genética da espécie humana pode
ser melhorada pelo uso da seleção artificial, uma ideia que ele chamou de eugenia: os genitores que
expressam características favoráveis deviam ser estimulados a ter famílias maiores (eugenia positiva), e os
genitores com características indesejáveis deveriam ser estimulados a não ter filhos (eugenia negativa). As
características que Galton achava favoráveis incluíam alta inteligência, altos níveis de conquistas,
criatividade artística e excelente saúde. As características que ele achava que deveriam ser selecionadas
contra incluíam baixa inteligência, doença mental, comportamento criminoso e alcoolismo.
63
Nos Estados Unidos, o movimento eugênico ganhou força durante a primeira parte do século XX,
especialmente a eugenia negativa. Em 1907, o estado do Indiana aprovou leis que determinavam a
esterilização dos indivíduos que eram "imbecis, idiotas, estupradores condenados e criminosos
contumazes". Em 1931 quase metade dos estados tinha adotado tais leis, e a esterilização compulsória foi
ampliada para incluir coisas como perversão sexual, vício de drogas, alcoolismo e epilepsia. Implícita nestas
leis de esterilização estava a ligação entre herança e comportamento. A esterilização dos retardados mentais
pode parecer-nos bárbara; entretanto, durante as décadas de 1920 e 1930, os procedimentos de esterilização
foram defendidos porque, diziam, as pessoas retardadas mentais eram genitores pobres e davam ambientes
pobres a seus filhos. Como não havia bons tratamentos com drogas que permitissem que as pessoas com distúrbios mentais tivessem vida normal e cuidassem dos filhos, foram internadas muito mais pessoas do que
hoje. A esterilização era considerada mais bondosa que o encarceramento.
Outro desdobramento do movimento eugênico durante a década de 1920 foram as leis de restrição à
imigração. Estas leis, motivadas amplamente por motivos económicos, favoreceram grupos étnicos
"geneticamente desejáveis" (principalmente os europeus do norte) e criaram severas restrições aos grupos
menos "geneticamente desejáveis" (os da área do Mediterrâneo, Europa Central, China). Estas leis eram
fundadas em "dados" errados e em fanatismo. Muitas delas permaneceram nos livros até a década de 1960.
O movimento eugênico adquiriu sua forma mais distorcida e perversa na Alemanha nazista. Entre 1930 e
1945, o regime de Hitler exterminou sistematicamente milhões de judeus, ciganos e outros, na tentativa de
"limpar" a Alemanha de material genético "inferior".
Nos Estados Unidos, na Inglaterra e em outras partes, a maioria dos geneticistas ficou assustada com o modo
pelo qual a ciência da genética, ainda muito jovem, estava sendo mal empregada. Não havia evidência sólida de que a
genética tivesse um papel na maioria das características humanas consideradas desejáveis ou indesejáveis. As pessoas
tinham informações leigas e as elevavam ao nível de fatos científicos (p. ex., Mr. Smith abusava da esposa e da criança;
seu filho também abusava da esposa e da criança; logo, abusar da esposa e da criança era um comportamento herdado).
Esta perversão da disposição da sociedade e da genética para aceitar isto sem crítica levou muitos geneticistas a evitar
o estudo da genética humana, com medo de ser considerados eugenicistas. O campo da genética humana sofreu
muito devido a estes abusos.
Hoje em dia, todas as universidades que recebem fundos federais para pesquisa estabeleceram comités que precisam
aprovar as propostas de pesquisa que envolvam questões humanas antes do início do trabalho. Além
disso,
o
National Institutes of Health do United States Department of Public Health criou comites que devem avaliar e aprovar
propostas de pesquisas genéticas em humanos antes de se iniciarem os trabalhos. Salvaguardas deste tipo devem nos
ajudar a evitar as pesquisas genéticas impróprias. A sociedade deve aprender as lições dos erros passados e caminhar
com otimismo para o século XXI. O Projeto Genoma Humano está produzindo uma grande quantidade de
informações novas sobre o genoma humano, e os genes que influenciam o risco de doenças genéticas estão sendo
64
rapidamente identificados. Com uma compreensão melhor sobre os genes e genomas, os cientistas devem ser capazes
de criar métodos eficazes para tratamento de muitas doenças humanas, como no passado já tinham para a
fenilcetonúria e para a hemofilia .
Pontos Importantes: O Projeto Genoma Humano está produzindo uma enorme quantidade de informações
sobre a genética de humanos. Se usadas com sabedoria, as novas informações prometem melhorar nossa
qualidade de vida. Entretanto, a disponibilidade desta informação também cria questionamentos morais, éticos e
legais que devem ser cuidadosamente avaliados pela sociedade.
REFERÊNCIAS BIBLIOGRÁFICAS
ALBERTS,B;
BRAY,D;
JOHNSON,A;
LEWIS,J;
RAFF,M;
ROBERTS,K;
WALTER,P
Fundamentos da Biologia Celular – Uma Introdução à Biologia Molecular da Célula. Ed. Artmed.
Porto Alegre, 1999, 757p.
Barros Veloso, A.J. 2003. Das ervilhas de Mendel à dupla hélice de Watson e Crick. Medicina
Interna. Vol.10, N. 3: 115-122.
Thompson & Thompson, Nussbaum RL, McInnes RR, Willard HF. (2002). Genética Médica. Ed.
Guanabara Koogan. 6ª edição.
Griffiths, A. J. F & Cols. (2001). Genética Moderna. Guanabara Koogan, 1ª edição.
Snustad, D. P. & Simons, M. J. (2001). Fundamentos de Genética. Guanabara Koogan, 2ª edição.
BROWN, T. A. Genética – um enfoque molecular. 3 ed. Editora: Guanabara Koogan, 1998.
L., GONIC; M., WHWWLIS. Introdução ilustrada à genética. São Paulo: Harbra, 1995.
CURTIS, Helena. Biologia. 2 ed. Rio de Janeiro: Guanabara Koogan,1977.
GARTNER, L.P. & HIATT, J. L. Tratado de Histologia - 1a edição, Guanabara Koogan, 1999,
426p.
JUNQUEIRA, L.C.U. & CARNEIRO, J. Histologia Básica 10a. edição. Ed. Guanabara Koogan,
2004, p.
65
SOBOTTA - Histologia. Atlas Colorido de Citologia, Histologia e Anatomia Microscópica Humana
-Guanabara Koogan, 1999.
SOARES, José Luis. Dicionário etimológico e circunstanciado de biologia. São Paulo: Scipione,
1993. 534 p.
66