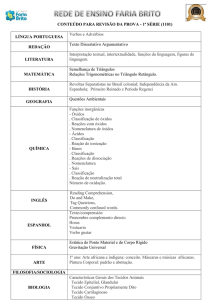
TERMOQUÍMICA
Introdução
Qualquer sistema num dado estado, possui uma certa quantidade de energia
(energia das ligações intra e inter moleculares, energia cinética correspondente ao
movimento das partículas, energia potencial gravítica, etc). Essa energia total de um
sistema chama-se energia interna, e representa-se por U.
Considere-se então uma variação da energia interna U= U2-U1, correspondente à
passagem de um estado inicial U1 a um estado final U2, e que pode conduzir a um
aumento ou a uma diminuição de energia interna do sistema. Dá-se assim uma troca de
energia com o exterior: ou uma transferência de energia do exterior para o sistema, U
0, ou do sistema para o exterior, U 0.
Essa transferência pode dar-se sob a forma de calor, q ou de trabalho, w, ou seja,
U= q + w
Por convenção, quando q 0, foi transferido calor para o sistema; quando q 0, o sistema
cedeu calor ao exterior. Do mesmo modo, se w 0, foi realizado trabalho sobre o sistema;
se w 0, o sistema realizou trabalho sobre o exterior.
A expressão U= q + w é a expressão matemática do 1º Princípio da
Termodinâmica: a energia não pode ser criada, nem destruída, ou de outro modo, a
energia do Universo é constante. Então, a variação da energia interna do sistema só
depende dos estados inicial e final e é independente do processo segundo o qual se deu
essa transformação, ou do caminho seguido entre os dois estados. Diz-se então que a
energia interna, tal como outras propriedades, é uma função de estado, é univocamente
definida pelo estado do sistema.
A maioria das reacções químicas ocorre em vaso aberto, quer dizer, a pressão
constante (já que a pressão atmosférica não variará significativamente durante o tempo de
reacção). Isso quer dizer que qualquer variação de volume corresponde a trabalho
realizado a pressão constante: o trabalho realizado pelo sistema será dado por
w = pv
*
Então a variação da energia interna de um sistema a sofrer uma reacção química a
pressão constante (só com variação de volume) é dada por
U = qp pv
em que qp é o calor adicionado ao sistema a pressão constante.
Assim, qp=U + pv = U2U1 + P (V2V1),
donde qp= (U2 + PV2 ) (U1 +PV1).
Já que P, V e U são funções de estado, definimos assim uma nova função de estado, a que
se chama entalpia, H.
H = U +PV
Como qp= (U2 + PV2 ) (U1 +PV1) = H2 H1 = H,
A variação de entalpia de um sistema, H, será então o calor adicionado ao sistema num
processo que ocorre a pressão constante e em que o único trabalho possível é o que
resulta da expansão contra a pressão atmosférica.
qp= H
*
Vamos supor que o sistema está contido num cilindro com uma secção de área A, e que o sistema se
expande e empurra a atmosfera de uma distância d. A variação de volume do sistema, v é igual a d×A. Da
Física sabemos que a energia usada para mover um objecto é igual a força F vezes o deslocamento do
objecto, d., F ×d. Mas F×d = F/A×(d×A) . Ora, F/A é igual à pressão e d×A = V. Sendo assim , a energia
usada pelo sistema para se expandir é Pv e como é trabalho realizado pelo sistema tem o sinal negativo.
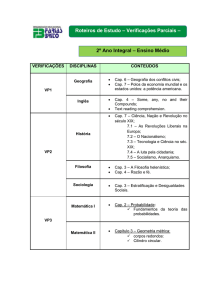
Ciclos de Born-Haber
Um ciclo de Born-Haber é um ciclo que estabelece relações entre várias grandezas
termodinâmicas. Baseia-se na lei de Hess ,"é possível calcular a variação de entalpia
associada a uma reacção química pela soma algébrica das variações de entalpia de outras
reacções químicas cujas equações, depois de somadas, dão a equação inicial”, ou seja,
numa consequência do 1º Princípio da Termodinâmica “a variação de entalpia entre um
estado inicial e um estado final é independente do percurso seguido entre os dois
estados" (esta propriedade é comum a todas as funções de estado, como é a entalpia, e
funções de estado são propriedades termodinâmicas que são univocamente definidas pelo
estado do sistema).
Considerem-se esquematicamente dois estados, um final e um inicial, que é
possível relacionar de forma directa através da variação de entalpia H1, ou então por
uma qualquer sucessão de estados intermédios separados pelas variações de entalpia
H2, H3 , ...Hn. Representa-se de forma abstracta um ciclo para n = 5.
H3
H2
Estado inicial
H4
H5
H1
Estado final
Segundo a Lei de Hess, H1 = - H2 + H3 - H4 + H5 , ou seja, o percurso
entre os estados inicial e final tanto pode ser "directamente" através de H1, como se
pode procurar um "caminho alternativo", que parta do mesmo estado inicial e chegue ao
mesmo final: neste caso é - H2 + H3 - H4 + H5 (os sinais menos aparecem quando o
sentido do percurso é contrário ao sentido das setas, quer dizer, quando nos estamos a
referir à reacção inversa daquela que está indicada no ciclo). Uma das utilidades destes
ciclos termodinâmicos é que, se apenas uma das variações de entalpia não for conhecida,
é possível calculá-la usando equações como esta. É assim que os ciclos de Born-Haber
têm sido usados, por exemplo, para calcular energias reticulares, entalpias de dissolução,
ou na previsão da estabilidade termodinâmica de um dado composto, através da sua
entalpia de formação.
Resumo de alguns conceitos, definições e convenções utilizados em ciclos
de Born-Haber
1. Grandezas relativas ao estado gasoso
Energia de dissociação ou de ligação
D(A-B) ≡ dissH, energia de dissociação E(AB), energia de ligação (é definida no
sentido da quebra das ligações): variação de entalpia associada ao processo de quebra
homolítica (i.e., cada átomo fica com um dos dois electrões que estabeleciam a ligação)
de uma mole de ligações, com reagentes e produtos no estado gasoso ideal, à pressão de 1
bar e á temperatura T = 298,15K.
B) D(A-B) diss
-
A(g) + B(g)
AB(g) E(A
Energia de ionização
EI : energia que é necessário fornecer para arrancar uma mole de electrões a uma mole de
átomos neutros, ou de iões, no estado gasoso ideal e fundamental, ficando os electrões
arrancados com uma energia cinética nula)
EI
A(g) A+(g) + e
Electroafinidade
EA : energia libertada quando se adiciona uma mole de electrões a uma mole de átomos
no estado gasoso ideal, para formar uma mole de iões mononegativos no estado
fundamental e também no estado gasoso ideal.
Por tradição, esta energia libertada é dada como positiva, i.e. os valores tabelados são
positivos, o que é contra a convenção termodinâmica; daí o sinal menos no processo
correspondente:
EA
A(g) + e A (g)
O processo inverso, +EA, corresponde à energia de ionização do ião A
EI ( A ) EA
A (g)
A(g) + e
2. Grandezas que envolvem (ou não) mudança de estado
Entalpia de formação padrão
fHº : variação de entalpia envolvida na formação de 1 mole de uma substância a partir
dos seus elementos constituintes nos respectivos estados padrão* a 298,15K.
*Estado padrão de uma substância define-se como a forma pura dessa substância à
pressão de 1 bar. Embora a temperatura não faça parte desta definição, os valores
tabelados costumam referir-se a 298,15K (25ºC).
Exemplo:
Δ Hº ( NaCl)
Na(c) + ½ Cl2(g) f
NaCl(c)
Por convenção, fHº (elemento no estado padrão) =0
Por exemplo, fHº (Cl2, g) = 0 , mas fHº(Cl, g) ≠0, porque o estado padrão do cloro (tal
como o hidrogénio, azoto, oxigénio, e os restantes halogéneos, flúor, bromo e iodo),
corresponde à molécula diatómica.
Entalpia de reacção
A variação de entalpia de uma reacção, rH, pode calcular-se a partir das entalpias de
formação padrão dos seus produtos e reagentes :
rH(variação da entalpia de uma reacção) =∑npfHº(produtos)∑ nrfHº(reagentes)
Por exemplo, consideremos a reacção
CH4(g) + 2O2(g) → CO2(g) + 2H2O(l)
rH = 2×fHº(H2O, l) + fHº(CO2, g) 2×fHº(O2, g) – fHº(CH4, g) = 2×fHº(H2O, l) +
fHº(CO2, g) – fHº(CH4, g)
Entalpia de atomização
atomH : variação de entalpia associada ao processo de cisão de todas as ligações de uma
mole de moléculas, ficando os átomos resultantes no estado gasoso ideal e com energia
cinética nula.
H
atom
A(g) + B(g) +..... + C(g)
AB...C(c,l ou g)
Entalpia de sublimação
subH : variação de entalpia associada à conversão de uma mole de composto ou
elemento cristalino em uma mole de composto ou elemento no estado gasoso.
Δ
H
A(c) sub
A(g)
Entalpia de vaporização
vapH : variação de entalpia associada ao processo de conversão de uma mole de um
composto ou elemento no estado líquido em uma mole de composto ou elemento no
estado gasoso.
Δ vap H
A(l) A(g)
Entalpia de dissolução
dissolH : variação de entalpia associada ao processo de dissolução de uma mole de um
composto iónico, ficando os seus iões solvatados em solução.
Δ
H
AB(c) dissol
A+(soln
) + B (soln)
Entalpia de
-se entalpia de hidratação)
solvH : variação de entalpia associada ao processo da passagem de uma mole de iões no
estado gasoso à solução.
Δ
H
A+(g) hid
A+(soln)
Energia reticular
Uret ≡ -U (energia que é necessário fornecer para separar 1 mole do cristal iónico nos
respectivos iões no estado gasoso perfeito)
U
AB(c) ret
A+(g) + B (g)
U é a energia das interacções electrostáticas e da repulsão das nuvens electrónicas numa
mole do cristal