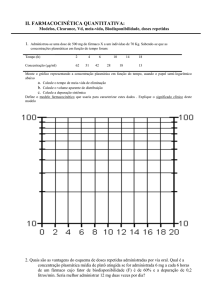
Turma Farmácia- 4º Termo
Profa. Dra. Milena Araújo Tonon
Farmacocinética : o que o organismo faz sobre a droga
Farmacodinâmica: o que a droga faz no organismo
Absorção: quanto do fármaco sai do seu local de administração para o interior da
corrente sanguínea
Biodisponibilidade: quantidade de droga que atinge seu local de ação ou um líquido
biológico a partir do qual o fármaco tem acesso ao seu local de ação
A absorção implica na passagem do fármaco através das membranas
Características importante do fármaco:
Tamanho , Forma farmacêutica (cápsula, comprimido, drágea), Grau de ionização,
Lipossolubilidade forma ionizada e não ionizada, Ligação a proteína tecidual
Maior absorção
Molécula não ionizada
A+ B -
AB
Maior absorção
Bases fracas
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Ácidos fracos
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Menor absorção
Administração
Absorção
Farmacocinética :
Fármaco na circulação sistêmica
Excretado
Distribuído
O que o organismo faz
sobre a droga
Biotransformado
Fármaco no sítio de ação
Farmacodinâmica:
Efeito farmacológico
Resposta clínica
O que a droga faz no
organismo
Distribuição
“Passagem de uma droga “livre” (farmacologicamente
ativa) da corrente circulatória para os tecidos”
Débito cardíaco, fluxo sanguíneo regional e o volume tecidual determinam a
taxa de liberação e a quantidade potencial de fármaco distribuída para os
tecidos
A extensão da distribuição depende:
• Irrigação dos tecidos
• Ligação a proteínas plasmáticas
•Permeabilidade através das membranas
Fluxo sangüíneo
A extensão da distribuição depende:
• Irrigação dos tecidos
• Ligação a proteínas plasmáticas
•Permeabilidade através das membranas
Ligação a proteínas plasmáticas
albumina (fármacos ácidos)
α1 glicoproteína ácida (fármacos básicos)
globulinas (α,β,γ)
β 1 lipoproteínas
O determinante mais importante da partição sangue:tecido é a ligação de um fármaco
a proteínas plasmáticas e macromoléculas teciduais
Somente o fármaco livre exerce atividade farmacológica
Sítios de ligação são inespecíficos e podem sofrer competição
- A substancia não-ligada constitui a forma farmacologicamente ativa.
A quantidade de um fármaco que se liga a proteínas vai depender de três fatores:
Concentração do fármaco livre
Afinidade do fármaco pelos locais de Ligação
Concentração de proteínas
O processo é não seletivo, saturável e não linear
Somente o fármaco livre exerce atividade farmacológica
A extensão da distribuição depende:
• Irrigação dos tecidos
• Ligação a proteínas plasmáticas
•Permeabilidade através das membranas
Difusão Pasiva
A molécula do fármaco atravessa a membrana em virtude da sua solubilidade na
dupla camada lipídica. Difusão a favor de um gradiente de concentração.
Quanto maior o coeficiente de partição maior a quantidade de fármaco na
membrana e mais rápida é a sua difusão
Hidrofobicidade da molécula
Maior absorção
forma não ionizada
A+ B -
AB
Maior absorção
Bases fracas
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Ácidos fracos
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Menor absorção
Fatores que interferem no padrão de
equilíbrio de distribuição entre os tecidos:
permeabilidade através das barreiras
pH e pKa (ionização)
lipossolubilidade
ligação no interior dos compartimentos
Distribuição das drogas em compartimentos especiais
Sistema nervoso central – Barreira hematoencefálica
As células endoteliais dos capilares do cérebro possuem junções de oclusão contínuas
As principais características das drogas que atravessam a barreira são:
Drogas apolares
Moléculas pequenas
Lipossolúveis
Elevado coeficiente de partição óleo/água
Distribuição das drogas em compartimentos especiais
Barreira placentária
A presença de fármacos podem causar anomalias congênitas. O plasma do feto é
ligeiramente mais ácido que o da mãe de forma que ocorre o sequestro de
substâncias básicas.
A proteína P atua como um transportador de exportação para limitar a exposição
fetal de substâncias tóxicas
As principais características das drogas que atravessam a barreira são:
Drogas apolares
Moléculas pequenas
Lipossolúveis
Elevado coeficiente de partição óleo/água
Volumes de Distribuição
Parâmetro farmacocinético que avalia a extensão da distribuição da
substância ativa, além do plasma
Volume real de distribuição: Volume anatômico acessível ao Fármaco.
Volume aparente de distribuição: Volume que o fármaco teria que se
dissolver para que sua concentração se igualasse à do plasma.
Vd= Q/ P
Onde:
Vd = volume aparente de distribuição
P = a concentração plasmática
Q = quantidade de fármaco administrada
Unidade : L
Músculo
Plasma
Fluido
extra
celular
Gordura
Fluido
intra
celular
• Drogas grandes, pesadas e com alta ligação a proteínas plasmáticas
(confinadas no compartimento plasmático) Ex. Heparina (Vd 0,05L/Kg muito
baixo)
•Drogas
com
baixa
lipossolubilidade
(atravessam
pouco
a
barreira)
principalmente compostos polares – (drogas distribuídas no compartimento
extracelular) Ex. Gentamicina (Vd 0,2l/Kg baixo)
•Drogas com relativa lipossolubilidade (alto poder de distribuição) – (distribuída
por toda água corporal ) Ex. (Etanol, diazepam Vd 0,55l/kg alto)
ABSORÇÃO
Tecidos: gordura, ossos
DISTRIBUIÇÃO
Depósito
Administração
com absorção
Droga livre
Local de ação do
efeito terapêutico
Droga + PP
Administração
sem absorção
Droga livre
Fígado:
Ativação, Inativação
BIOTRANSFORMAÇÃO
Local de ação do
efeito colateral
Rins, pulmões, etc
Emininação
Urina, fezes, ar
expirado etc.
EXCREÇÃO