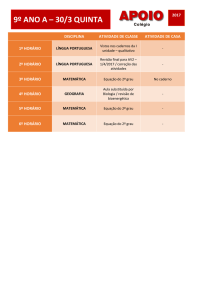
")
Catabolismo dos aminoácidos; Rui Fontes
Catabolismo dos aminoácidos 1 e 2 (duas aulas de grupo)
1-
No decurso do seu catabolismo os aminoácidos perdem os seus átomos de azoto que, na sua maioria, são
incorporados na ureia e excretados na urina. (i) A porção não azotada das moléculas dos aminoácidos (os
esqueletos carbonados) pode, em certos casos (a maioria), gerar intermediários do ciclo de Krebs ou da
glicólise. Nestes casos, os aminoácidos dizem-se glicogénicos porque, administrados a um animal em
jejum, podem, via gliconeogénese, formar glicose ou glicogénio. Quando se diz que um determinado
aminoácido é glicogénico quer-se apenas dizer que, potencialmente, o esqueleto carbonado deste
aminoácido pode, convertendo-se em glicose no fígado (e rim), ser indiretamente oxidado pelos tecidos do
organismo que consomem glicose. De facto, a ingestão de proteínas e a consequente absorção de
aminoácidos não provocam subida na glicemia porque um dos efeitos dos aminoácidos é a estimulação da
libertação de insulina nas células β pancreáticas o que estimula a oxidação da glicose e o seu
armazenamento na forma de glicogénio [1]. (ii) No caso da leucina os produtos do catabolismo são o
acetoacetato e o acetil-CoA e não se geram intermediários do ciclo de Krebs ou da glicólise; a leucina não
é um aminoácido glicogénico porque nenhum dos produtos formados a partir dela é substrato da
gliconeogénese e diz-se cetogénica porque o acetoacetato é um corpo cetónico e o acetil-CoA é o
precursor dos corpos cetónicos. O outro exemplo de aminoácido cetogénico é a lisina. Quando se diz que
um determinado aminoácido é cetogénico quer-se dizer que, potencialmente, o esqueleto carbonado deste
aminoácido pode, convertendo-se em acetoacetato e β-hidroxibutitato no fígado, ser indiretamente
oxidado pelos tecidos do organismo que consomem corpos cetónicos. (iii) Os aminoácidos que, no
decurso do seu catabolismo, se desdobram de tal forma que parte da molécula forma acetoacetato ou
acetil-CoA e a outra parte intermediários do ciclo de Krebs ou da glicólise costumam ser classificados
como simultaneamente glicogénicos e cetogénicos.
2-
Os intermediários do ciclo de Krebs podem (via fosfoenolpiruvato) gerar piruvato e este pode, por ação da
desidrogénase do piruvato, gerar acetil-CoA que é oxidado a CO2. O facto de os aminoácidos poderem,
no seu metabolismo, gerar piruvato, intermediários do ciclo de Krebs, acetoacetato e/ou acetil-CoA
permite compreender que, sendo oxidados a CO2, podem contribuir para a síntese de ATP sendo, a par
com os glicídeos e os lipídeos, "compostos energéticos". Nas dietas habituais na nossa cultura o valor
calórico das proteínas representa cerca de 15% do valor calórico total. Assim, embora a importância
“energética” dos aminoácidos seja menor que a dos glicídeos e lipídeos, o seu valor energético não é
negligenciável. É de notar que, embora os esqueletos carbonados dos aminoácidos sejam completamente
oxidados gerando CO2, o processo pode ser indireto: a maioria dos aminoácidos sofre catabolismo no
fígado onde o seu azoto origina ureia e o seu esqueleto carbonado acaba por originar glicogénio e glicose
(a maioria dos aminoácidos são glicogénicos). Para além do seu papel na síntese de (praticamente) toda a
ureia sintetizada no organismo, o fígado tem um importante papel no catabolismo do esqueleto carbonado
da maior parte dos aminoácidos estimando-se que metade da energia libertada nos processos oxidativos
que decorrem no fígado tenha origem na oxidação de aminoácidos [2]. Uma parte da importância do
fígado nos processos oxidativos dos aminoácidos decorre do facto de este órgão receber diretamente os
aminoácidos da dieta (via veia porta) captando e oxidando os que estão em excesso relativamente às
necessidades. O fígado também oxida glicose (e o seu próprio glicogénio) para fazer face às suas
necessidades energéticas e liberta glicose para o plasma (via gliconeogénese e via glicogenólise), mas uma
parte desta glicose teve origem no esqueleto carbonado dos aminoácidos. A ulterior oxidação desta glicose
nos diversos tecidos do organismo é também, em última análise, uma etapa do processo oxidativo dos
aminoácidos. Em termos médios, 1g de proteína, pode originar 0,6 g de glicose; se considerarmos que o
cérebro consome cerca de 100-120 g de glicose por dia, deve conclui-se que a ingestão de 100 g de
proteína (a ingestão “típica” diária numa dieta ocidental) pode contribuir para metade do consumo de
glicose pelo cérebro [2]. Em condições metabólicas extremas, como o jejum prolongado, o fígado
continua a produzir glicose e cerca de ¾ dessa glicose provém da conversão dos aminoácidos que são
libertados na proteólise endógena1.
3-
Embora a ureia e o amónio não resultem da oxidação dos esqueletos carbonados dos aminoácidos, o
processo de conversão dos aminoácidos em CO2 ou em glicose ou em corpos cetónicos é concomitante
1
O outro quarto provém da conversão do glicerol libertado na lipólise.
Página 1 de 11
Catabolismo dos aminoácidos; Rui Fontes
com a formação daqueles compostos de excreção. Por isso, a velocidade de degradação dos aminoácidos
no seu todo pode ser medida, medindo a velocidade de excreção dos compostos azotados na urina. O azoto
da ureia pode constituir entre 60% e 90% (a percentagem aumenta quando dieta é rica em proteínas) do
azoto urinário; a ureia, o amónio, a creatinina e o ácido úrico2 contêm mais de 95% do azoto urinário. Se
se considerarem períodos de tempo longos (vários dias) [3], o valor do azoto urinário presente na ureia e
no amónio é uma medida da velocidade de oxidação dos aminoácidos e pode servir para estimar a massa e
o valor energético dos aminoácidos que estão a ser oxidados.
4-
O catabolismo da alanina [3C,1N] é muito simples e envolve apenas a ação da transamínase da alanina
(ver Equação 1) que dá origem ao α-cetoácido correspondente, o piruvato [3C]. O piruvato é substrato da
gliconeogénese e pode, por isso, originar glicose. A alanina (cujo azoto constitui quase 10% do azoto
aminoacídico do plasma) é um veículo de transporte de azoto no plasma. No ciclo da alanina, o piruvato
formado na glicólise muscular aceita grupos amina de outros aminoácidos (ver Equação 1) convertendo-se
em alanina; a alanina sai dos músculos para o plasma sanguíneo; no fígado é captada e reconvertida em
piruvato (ver Equação 1); o piruvato, via gliconeogénese, gera glicose que pode voltar a ser oxidada no
músculo. Através da ação das enzimas da gliconeogénese hepática, glicólise muscular e transamínase da
alanina nos dois tecidos, o ciclo da alanina participa no transporte de azoto dos músculos para o fígado
(onde contribui para a formação de ureia), mas também permite que a glicose que, no músculo, foi apenas
oxidada a piruvato, possa ser regenerada no fígado. Do ponto de energético o ciclo da alanina, considerado
como um todo, consome ATP (consumo de 6 ligações ricas em energia e 2 NADH no fígado/molécula de
glicose formada e formação de 2 ligações ricas em energia e 2 NADH no músculo), mas permite poupar
glicose que é um importante substrato nos processos oxidativos cerebrais: tal como o ciclo do lactato (ou
de Cori), o ciclo da alanina também pode ser entendido como um processo de transferência de energia do
fígado para o músculo; as substâncias que estão a ser oxidadas no fígado permitem a formação de glicose,
cuja oxidação nos músculos, gera ATP. É, no entanto, de notar que os ciclos da alanina e do lactato não
permitem formar glicose de novo mas apenas recuperar como glicose a glicose que foi oxidada a piruvato
(nos músculos) ou cindida a lactato (nos eritrócitos e nos músculos). No cérebro, a glicose é oxidada a
CO2 e, num indivíduo em jejum prolongado (vários dias ou semanas), esta glicose provém
maioritariamente da conversão líquida dos aminoácidos endógenos em glicose.
Equação 1
5-
A asparagina [4C,2N], por ação da asparagínase, é hidrolisada gerando aspartato [4C,1N] e amónio
(ver Equação 2). O aspartato por transaminação (ver Equação 3) gera oxalacetato [4C] que é um
intermediário do ciclo de Krebs. No ciclo da ureia, o aspartato reage com a citrulina (sintétase do argininosuccinato) originando arginino-succinato. Nesta via metabólica o azoto do aspartato incorpora-se na ureia
e o esqueleto carbonato sai como fumarato [4C] que é também intermediário do ciclo de Krebs. Daqui se
pode concluir que a asparagina e o aspartato são aminoácidos glicogénicos.
Equação 2
Equação 3
6-
alanina + α-cetoglutarato ↔ piruvato + glutamato
asparagina + H2O → aspartato + NH4+
aspartato + α-cetoglutarato ↔ oxalacetato + glutamato
De forma semelhante ao caso da asparagina, a glutamina [5C,2N], por ação da glutamínase, dá origem a
glutamato (ver Equação 4) e o glutamato [5C,1N], por transaminação, gera o intermediário do ciclo de
Krebs α-cetoglutarato (ver Equação 5). No caso do glutamato, a formação do α-cetoglutarato [5C]
também pode resultar da ação da desidrogénase do glutamato (ver Equação 6). Os processos de hidrólise
do grupo amida da glutamina (ver Equação 4) e da asparagina (ver Equação 2) chamam-se,
frequentemente, de processos de desamidação porque o grupo químico onde ocorre a hidrólise é o grupo
amida presente nos carbonos 5 (caso da glutamina) e 4 (caso da asparagina). Os enterócitos têm
particular importância no catabolismo da glutamina (quer na que se forma a partir da hidrólise das
proteínas da dieta, quer na que se forma endogenamente).
2
A creatinina forma-se a partir da creatina e fosfocreatina que, por sua vez, se forma a partir da glicina, da arginina e da
metionina. A molécula da creatinina contém 3 átomos de azoto sendo que 1 provém diretamente da glicina e 2 da arginina.
O ácido úrico forma-se no catabolismo das purinas e a sua molécula contém 4 átomos de azoto: 2 provêm diretamente da
glutamina, 1 da glicina e o outro do aspartato.
Página 2 de 11
Catabolismo dos aminoácidos; Rui Fontes
Os enterócitos captam glutamina e uma parte da glutamina converte-se (via glutamato) em αcetoglutarato que, por sua vez, se converte em piruvato via ação de enzimas do ciclo de Krebs, da
carboxicínase do fosfoenolpiruvato e da cínase do piruvato; o piruvato, aceitando grupos azotados de
outros aminoácidos (transaminação) gera alanina que passa para a veia porta e é posteriormente
transformada em glicose (e ureia) no fígado. Assim, os enterócitos convertem três dos cinco carbonos da
glutamina em alanina (os outros dois convertem-se em CO2), mas não é este o único destino dos carbonos
da glutamina captada nos enterócitos. Um outro é a sua conversão em citrulina (via glutamato,
semialdeído do glutamato e ornitina) que passa para a circulação sanguínea e pode ser captada no fígado
ou no rim. Nestes processos o azoto do grupo amida da glutamina pode sair como amónio por ação da
glutamínase (ver Equação 4); este amónio também passa para a circulação sendo captado pelo fígado e aí
convertido em ureia. No entanto um outro destino do azoto do grupo amida da glutamina é a sua
incorporação no DNA dos enterócitos. Os enterócitos são células com uma taxa de multiplicação muito
elevada (a vida média dos enterócitos é de cerca de 5 dias) um processo que envolve a síntese de DNA e
dos nucleotídeos precursores. Nas vias de síntese dos nucleotídeos púricos e pirimídicos, a glutamina é
substrato de diversas enzimas que catalisam reações em que os carbonos da glutamina saem como
glutamato e o grupo amida se incorpora nos intermediários dessas vias.
Equação 4
Equação 5
Equação 6
7-
Numa reação fisiologicamente reversível a hidroxi-metil-transférase da serina pode catalisar a
interconversão da serina [3C,1N] e da glicina [2C,1N]; na reação também ocorre a interconversão do H4folato e do N5,N10-metileno H4-folato (ver Equação 7). A glicina pode ser oxidada (pelo NAD+) na ação
catalítica do complexo de clivagem de glicina; este complexo usa como aceitador de metilo o H4-folato e
na reação forma-se NADH, CO2, NH4+ e também N5,N10-metileno H4-folato (ver Equação 8). Assim,
por ação sequenciada da hidroxi-metil-transférase da serina e do complexo de clivagem de glicina, a serina
pode ser completamente oxidada formando CO2 e dois equivalentes de N5,N10-metileno H4-folato. (O
N5,N10-metileno H4-folato é substrato na síntese da timidina monofosfato e, portanto, importante para a
síntese de DNA.) Se atentarmos neste processo notaremos que a glicina (e indiretamente a serina) são
aminoácidos que podem ser oxidados a CO2 sem a intervenção de enzimas do ciclo de Krebs constituindo,
por isso, exceções ao processo oxidativo geral dos nutrientes.
Equação 7
Equação 8
8-
serina + H4-folato ↔ glicina + N5,N10-metileno H4-folato + H2O
glicina + NAD+ + H4-folato → CO2 + NH4+ + NADH + N5,N10-metileno H4-folato
A serina pode, por ação de outras enzimas, formar piruvato. Uma das vias metabólicas em que a serina
pode originar piruvato envolve, como primeiro passo, a ação de uma transamínase onde a serina perde o
grupo amina. Nesta via metabólica a serina origina, por transaminação, o 3-hidroxipiruvato (o α-cetoácido
correspondente à serina; ver Equação 9) que, através da ação de outras enzimas, acaba por gerar 2fosfoglicerato, um intermediário da glicólise e da gliconeogénese. O 2-fosfoglicerato pode converter-se
em glicose (gliconeogénese) ou originar piruvato e ser oxidado. Um outro processo, mais simples,
envolveria a ação da desidrátase da serina (ver Equação 10). Por ação sequenciada da hidroxi-metiltransférase da serina (ver Equação 7) e das enzimas que podem converter a serina em 2-fosfoglicerato ou
piruvato, a glicina pode também dar origem a piruvato.
Equação 9
Equação 10
9-
glutamina + H2O → glutamato + NH4+
glutamato + α-cetoácido ↔ α-cetoglutarato + α-aminoácido
glutamato + NAD+ + H2O → α-cetoglutarato + NADH + NH4+
serina + α-cetoglutarato ↔ 3-hidroxipiruvato + glutamato
serina → piruvato + NH4+
A cisteína [3C,1N,1S) contém um grupo tiol e as suas vias catabólicas são diversas e complexas. O grupo
tiol é oxidado gerando, maioritariamente, sulfato que é excretado na urina. De notar que o sulfato se
forma juntamente com os respetivos protões e que, portanto, o catabolismo da cisteína (e da metionina)
tende a acidificar o meio interno. O grupo amina da cisteína pode perder-se em reações de transaminação;
neste caso, o piruvato é também um dos produtos gerados no catabolismo da cisteína. Num outro
processo alternativo (quantitativamente menos relevante) forma-se taurina (C2,1N,1S) que, fazendo parte
dos ácidos biliares, é em última análise, excretada na urina. Na formação da taurina também ocorre
oxidação do grupo tiol mas, neste caso, o enxofre e o grupo amina mantêm-se ligados ao esqueleto
carbonado.
Página 3 de 11
Catabolismo dos aminoácidos; Rui Fontes
10-
A metionina [5C,1N,1S] é um aminoácido que contém um total de 5 carbonos e em que um deles (um
grupo metilo) se liga ao resto da cadeia por uma ligação sulfureto (CH3-S-CH2CH2CHNH2-COOH). No
processo catabólico, a metionina começa por reagir com o ATP gerando S-adenosil-metionina (ver
Equação 11). Um dos carbonos da metionina (o do metilo ligado ao enxofre) acaba transferido para
vários possíveis aceitadores (por ação de metil-transférases; ver Equação 12) formando-se um
intermediário contendo adenosina e homocisteína: a S-adenosil-homocisteína. A S-adenosil-homocisteína
é, de seguida, hidrolisada gerando a homocisteína (ver Equação 13). O átomo de enxofre da homocisteína
[4C,1N,1S] acaba transferido para a serina [3C,1N,1OH] que se converte em cisteína [3C,1N,1S]
enquanto o grupo azotado e os carbonos que pertenciam à homocisteína se libertam como NH4+ e αcetobutirato. Neste processo intervêm sequencialmente duas enzimas: a síntase da cistationina (ver
Equação 14) e a líase da cistationina (ver Equação 15). O α-cetobutirato (numa reação semelhante à que é
catalisada pela desidrogénase do piruvato) origina propionil-CoA (ver Equação 16) que, via metilmalonil-CoA, leva à formação de succinil-CoA que é um intermediário do ciclo de Krebs (ver Equações
17-19). A Equação 20 é a equação soma relativa ao processo de oxidação da metionina a succinil-CoA
(Equações 11-19). É de notar que, durante o catabolismo da metionina, o seu átomo de enxofre se converte
em enxofre da cisteína e que, portanto, este se perde maioritariamente como sulfato aquando do
catabolismo da cisteína. O grupo metilo é transferido para aceitadores de metilo. Se admitirmos que o CO2
que se perde na reação 16 é o mesmo que se incorpora durante a formação do succinil-CoA a partir do
propionil-CoA, poderemos também admitir que os outros 4 carbonos da metionina geram succinil-CoA. O
facto de o succinil-CoA ser um intermediário do ciclo de Krebs explica o caráter glicogénico da
metionina.
Equação 11
Equação 12
Equação 13
Equação 14
Equação 15
Equação 16
ATP + metionina → S-adenosil-metionina + Pi + PPi
S-adenosil-metionina + aceitador3 → S-adenosil-homocisteína + aceitador metilado
S-adenosil-homocisteína + H2O → homocisteína + adenosina
homocisteína + serina → cistationina
cistationina → cisteína + NH4+ + α-cetobutirato
α-cetobutirato + NAD+ + CoA → propionil-CoA + NADH + CO2
Equação 17
Equação 18
Equação 19
propionil-CoA + CO2 + ATP → D-metil-malonil-CoA + ADP + Pi
D-metil-malonil-CoA ↔ L-metil-malonil-CoA
L-metil-malonil-CoA ↔ succinil-CoA
Equação 20
metionina + 2 ATP + aceitador + H2O + serina + NAD+ + CoA →
succinil-CoA + cisteína + aceitador metilado + NH4+ + NADH + adenosina + PPi + 2 Pi + ADP
11-
A homocisteína, para além de poder reagir com a serina e formar cistationina (ver Equação 14), também
pode aceitar o grupo metilo do N5-metil-H4-folato regenerando-se metionina (síntase da metionina; ver
Equação 21). O N5-metil-H4-folato forma-se por redução (dependente do NADPH; ação da redútase do
N5,N10-metileno-H4-folato; ver Equação 22) do N5,N10-metileno-H4-folato (maioritariamente gerado no
catabolismo da serina e glicina; ver Equação 7 e Equação 8).
Equação 21
Equação 22
12-
N5-metil-H4-folato + homocisteína → H4-folato + metionina
N5,N10-metileno-H4-folato + NADPH → N5-metil-H4-folato + NADP+
No catabolismo da tirosina [9C,1N,1OH] a primeira reação é uma transaminação onde o grupo amina é
transferido para o α-cetoglutarato formando-se para-hidroxifenilpiruvato [9C] e glutamato (ver Equação
23). (O p-hidroxifenilpiruvato é o α-cetoácido correspondente à tirosina.) Em três reações sequenciais
catalisadas por duas oxigénases (um dos substratos é o O2) e uma isomérase, o p-hidroxifenilpiruvato dá
origem ao homogentisato [8C], ao maleilo-acetoacetato [8C] e ao fumaril-acetoacetato [8C] (ver Equação
24, Equação 25 e Equação 26). O fumaril-acetoacetato é, de seguida, hidrolisado (ver Equação 27)
3
Entre outros são aceitadores dos grupos metilo da S-adenosilmetionina a fosfatidil-etanolamina (formação de fosfatidilcolina), a noradrenalina (formação de adrenalina), o guanidoacetato (formação de creatina), resíduos de lisina e histidina em
proteínas e resíduos de nucleotídeos de ácidos nucleicos.
Página 4 de 11
Catabolismo dos aminoácidos; Rui Fontes
cindindo-se em fumarato [4C] e acetoacetato [4C]. A equação soma que descreve o catabolismo da
tirosina é a Equação 28. O facto de a cisão molecular do fumaril-acetoacetato gerar um intermediário do
ciclo de Krebs e um corpo cetónico explica a classificação da tirosina no grupo dos aminoácidos
“simultaneamente glicogénicos e cetogénicos”. Porque a fenilalanina [9C,1N] se converte em tirosina (ver
abaixo) o catabolismo da fenilalanina gera os mesmos produtos e a mesma classificação se aplica a este
aminoácido. A alcaptnúria é causada por uma deficiência congénita de uma enzima envolvida no
catabolismo da tirosina, a oxigénase do ácido homogentísico (ver Equação 25). Nesta doença, que não
põe em risco a vida, a acumulação de ácido homogentísico causa, como sinal mais relevante, uma urina
que escurece em contacto com o ar.
Equação 23
Equação 24
Equação 25
Equação 26
Equação 27
tirosina + α-cetoglutarato → p-hidroxifenilpiruvato + glutamato
p-hidroxifenilpiruvato + O2 → homogentisato + CO2
homogentisato + O2 → maleilo-acetoacetato
maleilo-acetoacetato ↔ fumaril-acetoacetato
fumaril-acetoacetato + H2O → fumarato + acetoacetato
Equação 28
tirosina + α-cetoglutarato + 2 O2 + H2O → fumarato + acetoacetato + glutamato + CO2
13-
A fenilalanina [9C,1N] converte-se em tirosina por ação de uma enzima hepática, a hidroxílase da
fenilalanina (diretamente dependente da tetrahidrobiopterina; ver Equação 29). Nesta reação a
fenilalanina e a tetrahidrobiopterina são oxidadas pelo oxigénio molecular originando, respetivamente,
tirosina e dihidrobiopterina; a regeneração da tetrahidrobiopterina ocorre por ação de uma redútase
dependente do NADPH (redútase da dihidrobiopterina; ver Equação 30). Quando uma destas enzimas
está deficiente ocorre a acumulação de fenilalanina que pode, por transaminação, gerar fenilpiruvato. Um
dos produtos a que o fenilpiruvato pode dar origem é o fenilacetato que surge na urina em quantidades
elevadas nesta situação patológica (designada por fenilcetonúria). A fenilcetonúria provoca lesões no
cérebro em desenvolvimento e, consequentemente, atraso mental grave. A causa das lesões cerebrais e do
atraso mental estará, provavelmente, relacionada com as concentrações elevadas de fenilalanina no plasma
sanguíneo e com a inibição (competitiva) que estas concentrações provocam na captação de outros
aminoácidos neutros (nomeadamente tirosina e triptofano) ao nível da barreira hematoencefálica [4]. Estas
complicações graves podem ser prevenidas com uma dieta pobre em fenilalanina durante, pelo menos, os
primeiros 6-8 anos de vida. Em Portugal colhe-se sangue a todos os bebés recém-nascidos sendo um dos
objetivos detetar (e tratar) precocemente esta doença. A doença é autossómica recessiva e tem uma
incidência relativamente elevada (1/13000 nascimentos). Desconhece-se o motivo da alta incidência das
mutações sendo legítimo especular que poderá estar relacionado com seleção positiva dos heterozigotos
em situações em que a fenilalanina escasseia (ou escasseava) na dieta.
Equação 29
Equação 30
14-
fenilalanina + tetrahidrobiopterina + O2 → tirosina + dihidrobiopterina + H2O
dihidrobiopterina + NADPH → tetrahidrobiopterina + NADP+
O catabolismo dos aminoácidos ramificados valina [5C,1N], isoleucina [6C,1N] e leucina [6C,1N] iniciase com a perda dos grupos α-amina em reações de transaminação (ver Equação 31, Equação 32 e Equação
33). Os esqueletos carbonados correspondentes formados são α-cetoácidos ramificados que, pela ação
catalítica de uma desidrogénase com atividade semelhante às desidrogénases que catalisam a oxidação
descarboxilativa do piruvato, α-cetoglutarato e α-cetobutirato, originam acis-CoA ramificados distintos
(ver Equação 34). Subsequentemente as vias metabólicas divergem. (1) No catabolismo da valina o
produto final é o succinil-CoA que se forma a partir do propionil-CoA via metil-malonil-CoA (ver
Equações 17-19). Assim, a valina leva à formação de um intermediário do ciclo de Krebs e é um
aminoácido glicogénico. (2) Um dos intermediários do catabolismo da leucina é o hidroxi-metil-glutarilCoA. Este composto é também um intermediário do ciclo de Lynen e a sua cisão (por ação da líase do
hidroxi-metil-glutaril-CoA) gera acetoacetato e a acetil-CoA. A classificação da leucina como
aminoácido cetogénico deriva do facto de um dos produtos do seu catabolismo ser o acetoacetato (um
corpo cetónico) e de a acetil-CoA (o outro produto), quando formado no fígado, poder também originar
acetoacetato (ciclo de Lynen). (3) No catabolismo da isoleucina, um dos intermediários (o α-metilacetoacetil-CoA) sofre cisão tiolítica originando acetil-CoA e propionil-CoA; num processo já referido a
propósito dos catabolismos da metionina e da valina, o propionil-CoA gera succinil-CoA (ver Equações
17-19). Assim, porque da cisão do intermediário α-metil-acetoacetil-CoA se gera acetil-CoA e um
Página 5 de 11
Catabolismo dos aminoácidos; Rui Fontes
composto (propionil-CoA) que é glicogénico, a isoleucina costuma classificar-se como um aminoácido
“simultaneamente glicogénico e cetogénico”.
Equação 31
Equação 32
Equação 33
Equação 34
leucina + α-cetoglutarato ↔ α-ceto-isocaproato + glutamato
isoleucina + α-cetoglutarato ↔ α-ceto-β-metil-valerato + glutamato
valina + α-cetoglutarato ↔ α-ceto-isovalerato + glutamato
α-cetoácido ramificado + CoA + NAD+ → acil-CoA ramificado + CO2 + NADH
Ao contrário do que acontece com a maioria dos outros aminoácidos que sofrem o seu catabolismo no
fígado, no intestino ou no rim, uma grande parte dos aminoácidos ramificados sofre catabolismo nos
músculos esqueléticos e cardíaco. Pelo menos a primeira reação em que estes aminoácidos intervêm (a
de transaminação; ver Equação 31, Equação 32 e Equação 33) é um processo que é mais ativo nos
músculos. O azoto do grupo amina destes aminoácidos sai dos músculos incorporado na alanina e na
glutamina4.
15-
(1) O catabolismo da arginina [6C,4N] está intimamente associado ao seu papel como intermediário do
ciclo da ureia. Neste ciclo, a hidrólise da arginina (pela argínase) leva à formação de ornitina [5C,2N] e
ureia [(NH2)2CO]. A ornitina contém um grupo amina no carbono 5 e é substrato de uma transamínase; no
processo catalítico, o grupo amina converte-se num grupo aldeído formando-se o semialdeído do
glutamato (ver Equação 35). A oxidação do grupo aldeído do semialdeído do glutamato leva à formação
de glutamato que, como já referido, se pode converter em α-cetoglutarato (ver Equação 5 e Equação 6).
(2) O catabolismo da prolina [5C,1N] está relacionado com o da arginina na medida em que um
intermediário comum é o semialdeído do glutamato. Assim, quer a arginina, quer a prolina são
aminoácidos glicogénicos.
Equação 35
16-
O primeiro passo no catabolismo da histidina [6C,3N] é a sua desaminação que, ao contrário do que é
mais comum, é catalisada por uma líase, a histídase (ver Equação 36). O urocanato formado ainda contém
dois átomos de azoto que são constituintes do anel imidazol. A via catabólica leva, a dado passo do
processo, à formação do formimino-glutamato (Figlu); o segundo átomo de azoto (e um dos carbonos) é
transferido desse intermediário para o H4folato formando-se, nessa reação, N5-formimino-H4folato e
glutamato (ver Equação 37). O azoto do grupo formimino do N5-formimino-H4folato perde-se na forma
de amónio gerando N5,N10-metenil-H4folato (ver Equação 38) que intervém na síntese das bases púricas.
O glutamato, como já referido, pode gerar α-cetoglutarato e, por isso, a histidina é um aminoácido
glicogénico.
Equação 36
Equação 37
Equação 38
17-
α-cetoácido + ornitina ↔ α-aminoácido + semialdeído do glutamato
histidina → urocanato + NH4+
formimino-glutamato (Figlu) + H4folato → N5-formimino-H4folato + glutamato
N5-formimino-H4folato → N5,N10-metenil-H4-folato + NH4+
O processo catabólico da treonina [4C,1N,1OH] nos seres humanos é incerto [3-4], mas sabe-se que a
treonina não é substrato em reações de transaminação. Uma das vias metabólicas possíveis envolve a cisão
da molécula de treonina: dois dos seus carbonos e o azoto dariam origem a glicina (que pode gerar
piruvato) e os outros dois carbonos ao resíduo acetato da acetil-CoA. A reação envolveria a ação catalítica
de um complexo enzimático designado por complexo de clivagem da treonina (ver Equação 39). Numa
outra via metabólica alternativa a treonina sofre desaminação por ação catalítica da desidrátase da serina
4
Embora seja controverso, admite-se que na formação do esqueleto carbonado da glutamina no músculo possam intervir
conjuntamente os produtos de todos os aminoácidos ramificados. No ciclo de Krebs, o succinato (formado a partir da valina
e isoleucina) pode gerar oxalacetato que, reagindo com a acetil-CoA (eventualmente proveniente do catabolismo da
isoleucina e leucina), pode formar citrato e sequencialmente α-cetoglutarato. O α-cetoglutarato poderá aceitar grupos amina
na primeira reação do catabolismo dos aminoácidos ramificados (ver Equação 31, Equação 32 e Equação 33) formando
glutamato. O glutamato pode gerar glutamina (ação catalítica da sintétase da glutamina: glutamato + NH3 + ATP →
glutamina + ADP + Pi) incorporando NH3 formado no catabolismo de outros aminoácidos. A glutamina é o aminoácido
mais abundante no plasma sanguíneo constituindo por si só quase 1/3 do azoto aminoacídico do plasma e, conjuntamente
com a alanina (ciclo da alanina), um veículo de transporte de azoto dos músculos para o fígado.
Página 6 de 11
Catabolismo dos aminoácidos; Rui Fontes
originando-se NH4 e α-cetobutirato (ver Equação 40). O processo de conversão do α-cetobutirato em
succinil-CoA (intermediário do ciclo de Krebs) já foi referido (ver equações 16-19). Devido à existência
das dúvidas apontadas a treonina é, às vezes, classificada como glicogénica e, outras, como
simultaneamente glicogénica e cetogénica.
+
Equação 39
Equação 40
18-
treonina + CoA + NAD+→ acetil-CoA + glicina + NADH
treonina → α-cetobutirato + NH4+
No catabolismo da lisina [6C,2N], o grupo amina terminal é transferido para o α-cetoglutarato gerando-se
glutamato e o semialdeído do α-aminoadipato. O processo envolve oxiredútases e poderia, em última
análise, ser entendido como “uma transaminação”; no entanto, não é catalisado por uma transamínase do
tipo das que operam no caso dos outros aminoácidos. O semialdeído do α-aminoadipato é oxidado a αaminoadipato que já é substrato de uma transamínase clássica; é na ação catalítica desta transamínase que
o grupo α-amina se perde para o α-cetoglutarato (Equação 41). O produto final do catabolismo da lisina é
o acetoacetil-CoA (intermediário da síntese dos corpos cetónicos) e por isso a lisina é classificada como
um aminoácido cetogénico. O acetoacetil-CoA pode ser cindido pela ação da tiólase gerando acetil-CoA.
Equação 41
α-aminoadipato + α-cetoglutarato → α-cetoadipato + glutamato
19-
A primeira enzima envolvida no catabolismo do triptofano [11C,2N] é uma oxigénase cuja ação leva à
rotura do anel indole. Embora o triptofano possa gerar acetoacetil-CoA, na via catabólica do triptofano
ocorrem reações laterais que podem levar à formação de produtos alternativos. A cinurenina é o primeiro
intermediário da via catabólica que pode ser substrato em processos reativos alternativos. Num destes
processos a cinurenina leva à formação de 3-hidroxiantranilato e alanina; no outro gera-se ácido
cinurénico que é excretado na urina. Porque, no seu catabolismo, pode gerar alanina (que gera piruvato) e
acetoacetil-CoA, o triptofano é classificado como um aminoácido simultaneamente glicogénico e
cetogénico. Na via metabólica que converte o 3-hidroxiantranilato em acetoacetil-CoA forma-se um
intermediário que, em reações alternativas, pode gerar o ribonucleotídeo do ácido nicotínico, precursor na
síntese do NAD+ e do NADP+.
20-
De acordo com o critério referido no ponto 1 seriam classificados como aminoácidos cetogénicos a
leucina e a lisina. A tirosina e a fenilalanina (que originam fumarato e acetoacetato), o triptofano (que
origina alanina e acetocetil-CoA) e a isoleucina (que origina succinil-CoA e acetil-CoA) seriam
classificados como simultaneamente cetogénicos e glicogénicos. Seriam aminoácidos glicogénicos: a
asparagina e o aspartato (que originam oxalacetato ou fumarato), a glutamina, o glutamato, a arginina, a
ornitina, a prolina e a histidina (que originam α-cetoglutarato), a alanina, a serina, a glicina e a cisteína
(que originam piruvato) e a metionina e a valina (que originam succinil-CoA). De facto, mesmo durante o
período absortivo, uma parte dos hepatócitos (os hepatócitos peri-portais) continua a formar glicose-6fosfato a partir dos aminoácidos glicogénicos (e simultaneamente glicogénicos e cetogénicos) absorvidos,
armazenando os carbonos correspondentes a estes aminoácidos na forma de glicogénio; é a chamada
glicogénese indireta [5]. Como já referido, este glicogénio pode, via glicogenólise, fornecer glicose-6fosfato para oxidação nas mesmas células ou, via glicose-6-fosfátase, libertar glicose para o plasma
sanguíneo.
21-
Com as exceções da glicina e da serina (via glicina) que podem ser completamente oxidados a CO2 pela
ação do complexo de clivagem da glicina, a oxidação completa dos aminoácidos implica, mesmo no caso
dos aminoácidos glicogénicos e dos simultaneamente glicogénicos e cetogénicos, a formação de acetilCoA e o envolvimento das enzimas do ciclo de Krebs. Quando um determinado aminoácido é oxidado
de forma completa num órgão em que não há gliconeogénese, o intermediário do ciclo de Krebs formado
no catabolismo desse aminoácido é oxidado via conversão desse intermediário em oxalacetato (ciclo de
Krebs) e posterior conversão deste em fosfoenolpiruvato (carboxicínase do fosfoenolpiruvato), piruvato
(cínase do piruvato) e acetil-CoA (desidrogénase do piruvato)5.
5
De facto, é possível que esta via direta de oxidação dos aminoácidos glicogénicos possa também ocorrer no fígado quando,
durante a fase absortiva, há elevadas concentrações de aminoácidos na veia porta e estes são (com exceção dos aminoácidos
ramificados) maioritariamente captados pelo fígado. Admite-se assim que, quando a refeição é rica em proteínas e uma
parte dos aminoácidos estão, no fígado, a ser usados como substratos na síntese de glicogénio hepático (via gliconeogénese,
Página 7 de 11
Catabolismo dos aminoácidos; Rui Fontes
22-
No metabolismo da serina, da glicina, da histidina e da metionina intervêm derivados do folato.
(a) No catabolismo da serina e da glicina o H4-folato é aceitador de unidades monocarbonadas formandose o N5,N10-metileno-H4-folato (ver Equação 7 e Equação 8) que, por sua vez, é dador de unidades
monocarbonadas à 2'-desoxi-uridina monofosfato (2'd-UMP) sintetizando-se timidina monofosfato
(TMP); ver Equação 42). O dihidrofolato (H2-folato) que se forma no processo é reduzido a H4-folato
pela redútase do dihidrofolato (ver Equação 43).
Equação 42
Equação 43
N5,N10-metileno-H4-folato + 2'-desoxi-uridina monofosfato →
H2-folato + timidina monofosfato
H2-folato + NADPH → H4-folato + NADP+
(b) O carbono do grupo metileno (N5 - CH2 – N10) do N5,N10-metileno-H4-folato tem número de
oxidação zero. Numa reação de redução catalisada pela redútase do N5,N10-metileno-H4-folato este
composto dá origem ao N5-metil-H4-folato (ver Equação 22) que é capaz de transferir o grupo metilo
(N5-CH3; o carbono do grupo metilo tem número de oxidação –2) para a homocisteína e formar metionina
(síntase da metionina: ver Equação 21; esta síntase tem como cofactor a vitamina B12). Assim, via
metilação do H4-folato pela glicina ou pela serina e subsequente redução do metileno-H4-folato a metilH4-folato forma-se o dador de metilo para a regeneração da metionina. A metionina “ativada” (Sadenosil-metionina; ver Equação 11) é dador de metilos aquando da síntese de variados compostos como,
por exemplo, a fosfatidil-colina a partir de fosfatidil-etanolamina e a adrenalina a partir da noradrenalina
(ver Equação 12). Nestas reações, em que intervém como dador de metilo a S-adenosil-metionina, formase a S-adenosil-homocisteína que, ao ser hidrolisada, gera homocisteína (ver Equação 13). Como já
referido, a homocisteína pode ser metilada pelo N5-metil-H4-folato regenerando a metionina (ver Equação
21).
(c) O N5,N10-metileno-H4-folato (formado no catabolismo da serina e glicina; ver Equação 7 e Equação
8) pode ser oxidado por desidrogénases do N5,N10-metileno-H4-folato e gerar N5,N10-metenilo-H4folato (ver Equação 44). O N5,N10-metenilo-H4-folato (assim como a sua forma hidratada N10-formilH4-folato que resulta de hidrólise intramolecular) é dador de unidades monocarbonadas durante o
processo de síntese dos nucleotídeos púricos. O carbono do grupo metenilo do N5,N10-metenilo-H4folato (N5 – CH = N10) tem número de oxidação +2. O carbono do grupo formimino do N5-formiminoH4-folato (N5 – CH = NH) e o do grupo formilo do N10-formil-H4-folato (O = CH - N10) também têm
número de oxidação +2. O N5-formimino-H4-folato pode (por desaminação) dar origem ao N5,N10metenilo-H4-folato e este (por hidratação) pode originar o N10-formil-H4-folato. O N5-formimino-H4folato forma-se durante o catabolismo da histidina aquando da transferência do grupo formimino do
formimino-glutamato (Figlu) para o H4-folato (ver Equação 37).
Equação 44
23-
N5,N10-metileno-H4-folato + NADP+ ou NAD+ →
N5,N10-metenilo-H4-folato + NADPH ou NADH
No seu processo catabólico, a perda dos átomos de azoto dos aminoácidos pode ocorrer em diferentes
tipos de reações.
(1) Nos casos da glutamina e da asparagina o azoto do grupo amida sai como NH4+ por hidrólise e o
processo chama-se desamidação (ver Equação 2 e Equação 4).
(2) O grupo α-amina do glutamato e da glicina pode perder-se por desaminação oxidativa formando-se
também NH4+. No primeiro caso está envolvida a desidrogénase do glutamato e no segundo a enzima de
clivagem da glicina (ver Equação 6 e Equação 8). (No caso da serina, a possibilidade de se poder
converter em glicina explica que a desaminação oxidativa também possa estar indiretamente implicada na
perda do seu grupo amina.)
um processo designado de glicogénese indireta), o ATP consumido no processo provenha da oxidação da parte restante dos
aminoácidos que estão a ser diretamente oxidados via conversão em acetil-CoA [Newsholme e Leech (2010) Functional
Biochemistry in Health and Disease]. De facto, se pensarmos no processo na sua globalidade, a existência desta via direta de
oxidação dos aminoácidos é indistinguível de um processo em que o glicogénio formado nos hepatócitos a partir de
aminoácidos sofre oxidação completa nos mesmos hepatócitos onde se forma; a conversão do fosfoenolpiruvato em
glicogénio e deste em fosfoenolpiruvato seria um ciclo de substrato (em sentido lato).
Página 8 de 11
Catabolismo dos aminoácidos; Rui Fontes
(3) No caso do glutamato um processo alternativo para a perda do grupo α-amina é o envolvimento de
reações de transaminação em que diversos α-cetoácidos podem funcionar como aceitadores do grupo
amina do glutamato. As reações de transaminação são catalisadas por transamínases e a maioria dos
aminoácidos pode perder o grupo α-amina em reações catalisadas por transamínases em que os
aminoácidos funcionam como dadores do grupo amina ao α-cetoglutarato. Para além do caso do
glutamato são especialmente relevantes para a perda do seu grupo amina os processos de transaminação da
alanina (ver Equação 1), do aspartato (ver Equação 3), da serina (ver Equação 9), tirosina (ver Equação
23) e dos aminoácidos ramificados (ver Equação 31, Equação 32 e Equação 33). A transferência direta do
grupo α-amina do aminoácido não transformado em reações catalisadas por transamínases não ocorre
normalmente (ou não parece ter importância fisiológica) no catabolismo da glicina, da treonina, da
metionina, da lisina, da arginina, da histidina, da prolina, do triptofano e da fenilalanina. Contudo, é de
salientar, que a análise das vias metabólicas permite compreender a importância deste tipo de reações na
perda dos grupos α-amina de muitos dos aminoácidos acima referidos: nos casos da lisina, da arginina, da
prolina, do triptofano, da fenilalanina e cisteína são catabolitos α-aminados destes aminoácidos que
perdem o grupo amina em reações de transaminação clássicas. Os grupos amina terminais da ornitina
(formada a partir da arginina) e da lisina também se perdem em reações que também se podem designar
por "reações de transaminação". No caso da ornitina a transamínase envolvida na perda do grupo 5-amina
é semelhante às outras transamínases. No caso da lisina a reação de transferência do grupo 6-amina para o
α-cetoglutarato envolve uma oxiredútase e o termo “transaminação” só em sentido lato pode ser aplicado..
(4) Nos casos da serina, da treonina e da histidina a perda do grupo α-amina pode ser catalisado por líases
(a desidrátase da serina e a histídase são líases; ver Equação 10, Equação 40 e Equação 36). Um dos
intermediários no catabolismo da metionina, a cistationina, também perde o grupo α-amina por ação de
uma líase (a cistationínase é uma líase; ver Equação 15). (No caso da glicina, a possibilidade de se poder
converter em serina explica que a transaminação também possa estar indiretamente implicada na perda do
seu grupo amina.)
(5) A histidina contém, no anel imidazol, dois azotos sendo que um deles gera o grupo α-amina do
glutamato; o outro sai ligado a uma unidade monocarbonada gerando N5-formimino-H4-folato que por
desaminação não hidrolítica (uma líase; ver Equação 38) dá origem a amónio.
(6) A maior parte do azoto do anel indole do triptofano perde-se como amónio por desaminação oxidativa
de um intermediário do processo catabólico.
(7) A arginina contém quatro azotos; dois dos azotos perdem-se na forma de ureia por ação hidrolítica da
argínase. Os outros dois azotos ficam incorporados na ornitina que pode perder o azoto 5-amina por
transaminação gerando simultaneamente semialdeído do glutamato (ver Equação 35); por sua vez, o
semialdeído do glutamato pode ser oxidado a glutamato.
24-
A esmagadora maioria dos átomos de azoto dos aminoácidos acaba por ser excretado na urina na forma de
ureia. No ciclo da ureia, a arginina [6C,4N] gera diretamente ureia quando se cinde por ação hidrolítica da
argínase em ornitina [5C,2N] e ureia [1C,2N]. O azoto dos outros aminoácidos, quer diretamente
(desamidação hidrolítica, desaminação oxidativa ou ação de líases), quer indiretamente (via
transaminações com o α-cetoglutarato e subsequente desaminação oxidativa do glutamato formado (ver
Equação 6), origina NH4+ que é precursor de um dos dois azotos da ureia. O outro azoto da ureia provém
diretamente do aspartato mas, porque o azoto de todos os aminoácidos pode ser incorporado no grupo
amina do glutamato e este pode transaminar com o oxalacetato para formar aspartato (ver Equação 3),
compreende-se que também este “segundo azoto” pode, em última análise, provir de todos os
aminoácidos.
1. Gannon, M. C. & Nuttall, F. Q. (2010) Amino acid ingestion and glucose metabolism--a review, IUBMB Life. 62, 660-8.
2. Frayn, K. N. (2003) Metabolic regulation. A human perspective., 2nd edn, Blackwell Science, Oxford.
3. Matthews, D. E. (2006) Proteins and aminoacids in Modern Nutrition in Health and Disease (Shils, M. E., ed) pp. 23-61, Lippincott,
Phyladelphia.
4. van Spronsen, F. J., Hoeksma, M. & Reijngoud, D. J. (2009) Brain dysfunction in phenylketonuria: is phenylalanine toxicity the only
possible cause?, J Inherit Metab Dis. 32, 46-51.
5. Stipanuk, M. H. (2006) Biochemical, Physiological, Molecular Aspects of Human Nutrition, 2nd edn, Sunders, Elsevier., St. Louis.
Página 9 de 11
Catabolismo dos aminoácidos; Rui Fontes
Página 10 de 11
Catabolismo dos aminoácidos; Rui Fontes
Página 11 de 11
")