Fundação Oswaldo Cruz
Instituto Fernandes Figueira
Pós-Graduação em Saúde da Criança e da Mulher
PERFIL CLÍNICO E LABORATORIAL DE PACIENTES COM SUSPEITA DE
IMUNODEFICIÊNCIA PRIMÁRIA ATENDIDOS EM UM SERVIÇO DE
REFERÊNCIA DO RIO DE JANEIRO
Aniela Bonorino Xexéo Castelo Branco
Rio de Janeiro
Agosto de 2008
Fundação Oswaldo Cruz
Instituto Fernandes Figueira
Pós-Graduação em Saúde da Criança e da Mulher
PERFIL CLÍNICO E LABORATORIAL DE PACIENTES COM SUSPEITA DE
IMUNODEFICIÊNCIA PRIMÁRIA ATENDIDOS EM UM SERVIÇO DE
REFERÊNCIA DO RIO DE JANEIRO
Aniela Bonorino Xexéo Castelo Branco
Dissertação apresentada à PósGraduação em Saúde da
Criança e da Mulher, como parte
dos requisitos para obtenção do
título de Mestre em Ciências da
Saúde
Maria Ignez Capella Gaspar Elsas
José Marcos Telles da Cunha
Rio de Janeiro
Agosto de 2008
FICHA CATALOGRÁFICA NA FONTE
INSTITUTO DE COMUNICAÇÃO E INFORMAÇÃO
CIENTÍFICA E TECNOLÓGICA EM SAÚDE
BIBLIOTECA DA SAÚDE DA MULHER E DA CRIANÇA
C348
Castelo Branco, Aniela Bonorino Xexéo
Perfil clínico e laboratorial de pacientes com suspeita de
imunodeficiência primária atendidos em um serviço de referência
do Rio de Janeiro. / Aniela Bonorino Xexéo Castelo Branco. – 2008.
150 f.
Dissertação ( Mestrado em Saúde da Criança e da Mulher ) Instituto Fernandes Figueira , Rio de Janeiro , RJ , 2008 .
Orientador : Maria Ignez Cepella Gaspar Elsas
Co-orientador: José Marcos Telles da Cunha
Bibliografia: f. 107 -111
1. Síndromes de Imunodeficiência. 2. Pediatria. 3. Alergia e
Imunologia. 4. Infecção. 5. Saúde Pública. Título.
CDD - 22ª ed. 616.979
Dedico
ao meu marido Henderson e
à minha filha Amanda.
AGRADECIMENTOS
Ao Instituto Fernandes Figueira, ao Instituto de Puericultura e Pediatria
da UFRJ e aos seus respectivos corpos docente e técnico-administrativo, que
viabilizaram a obtenção dos dados para esta dissertação de Mestrado.
Aos meus orientadores José Marcos e Maria Ignez pela dedicação,
paciência ao longo do desenvolvimento deste trabalho.
Aos professores e coordenadores da Pós-Graduação em Saúde da
Mulher e da Criança do IFF/Fiocruz, pelos ensinamentos prestados.
Aos membros da banca examinadora
contribuições para a melhoria de nosso trabalho.
pela
disponibilidade
e
Aos colegas do Serviço de Pediatria e Alergia do HSE-RJ, pela
colaboração indispensável durante a realização deste estudo.
Aos meus pais Xexéo e Luana pelo amor e carinho com que me criaram
e por tudo que me ensinaram e continuam ensinando todos esses anos.
Aos meus amigos e colegas do mestrado, em especial à minha amiga e
sócia Fernanda pela ajuda na coleta de dados.
Ao meu irmão Geraldo pela ajuda com a análise dos dados e à minha
irmã Valéria pela ajuda cuidando da minha filha.
Aos meus sogros Castelo e Maria do Carmo pela ajuda cuidando da
minha filha.
Ao meu marido Henderson pela grande ajuda com a análise dos dados e
pelo apoio durante todo o período do mestrado
RESUMO
Objetivos: descrever o perfil clínico e laboratorial dos pacientes com suspeita
de imunodeficiência primária (IDP) atendidos no serviço de alergia e imunologia
pediátrica do IPPMG , verificar se o formulário de consulta de primeira vez
utilizado no serviço acelera a definição do diagnóstico e propor um novo
formato de formulário para a coleta de dados na consulta de primeira vez e no
acompanhamento dos pacientes no serviço.
Metodologia: estudo transversal descritivo retrospectivo de todas as crianças
atendidas no ambulatório de alergia e imunologia do IPPMG/UFRJ, no período
de janeiro de 2006 a junho de 2008. A coleta de dados foi realizada através de
revisão de prontuário e do formulário de primeira vez do serviço. Foram
excluídos pacientes cujos prontuários ou formulários de primeira vez não
puderam ser acessados.
Resultados: foram incluídos 181 pacientes, 81 pacientes (44,8%) do sexo
feminino e 100 pacientes (55,2%) do sexo masculino. Cinco pacientes (3%)
relataram consangüinidade e 28% tinham história familiar de IDP positiva. A
idade dos primeiros sintomas variou do nascimento a 132 meses com média de
21,8 (+ 30) e mediana de 8 meses. A idade na primeira visita variou de 0,2 a
203,51 meses com média de 67,52 (+49,43) e mediana de 59,21 meses. A
ocorrência infecções de repetição foi o principal motivo de encaminhamento
com 135 casos (75%). O principal sítio de infecção encontrado foi pneumonia,
com 119 casos (66%). Relataram internações 122 pacientes (67%), com média
de número de internações de 3.25 (+3.03) e mediana de 2 internações. A
média de dias internado foi de 12.65 (+12.37) com mediana de 9,5. Ao todo
foram solicitados 978 exames. Destes, 272 (28%) não foram realizados ou o
resultado não foi encontrado. A maioria dos diagnósticos pertenceu ao grupo
de imunodeficiências predominantemente de anticorpos com 15 casos (19%),
seguido de defeitos congênitos de fagócitos com 6 casos (8%), outras
síndromes bem definidas com 4 casos (5%), doenças de desregulação imune e
imunodeficiências combinadas, ambas com 2 casos (3%) e deficiências de
complemento com 1 caso (1%) . O grupo outros totalizou 9 casos (11%) e 3
casos (4%) tiveram diagnóstico de imunodeficiência secundária. A análise
comparativa do tempo decorrido desde a primeira visita até o diagnóstico a
partir do emprego ou não do formulário de primeira vez não foi estatisticamente
significativa (p > 0,93). A antibioticoterapia profilática foi indicada para 58 casos
(32%), a conduta foi expectante em 61 casos (34%) e gamaglobulina humana
(IVIG) foi indicada para 14 pacientes (8%). Três pacientes fizeram transplante
de células-tronco hematopoiéticas. A maioria dos pacientes manteve o
seguimento (54%), quatro pacientes (2%) foram à óbito e 15% dos casos não
havia registro de consulta de seguimento ou óbito, sendo caracterizado como
abandono de acompanhamento.
Palavras-chave: síndromes de imunodeficiência, pediatria, alergia e
imunologia, infecção, saúde pública.
ABSTRACT
Objectives: to describe clinical and laboratorial features of patients with
suspected primary immunodeficiency disease (PID) attended at the Allergy and
Immunology outpatient clinic at IPPMG, to assess if the first evaluation form
used at the service accelerates the definition of the diagnosis and propose a
new form for data collection at both first-time consultation and follow-up visits of
patients at the service.
Methodology: Retrospective analysis of all children who have attended the
Allergy and Immunology clinic at IPPMG, from January 2006 to June 2008. The
study was performed through review of conventional medical records and the
form used at the first consultation. Patients whose records or forms could not be
reached were excluded.
Results: 181 patients were included, 81 patients (44.8%) females and 100
patients (55.2%) males. Five patients (3%) reported consanguinity and 28% had
a positive family history of PID. The age of first symptoms ranged from birth 132
months with an average of 21.8 (30) and a median of 8 months. The age at first
visit ranged from 0.2 to 203.51 months with an average of 67.52 (49.43) and a
median of 59.21 months. The occurrence of repeated infections was the main
reason for referral with 135 cases (75%). The primary site of infection was
pneumonia, with 119 cases (66%). Admissions were reported for 122 patients
(67%), with a mean number of admissions of 3.25 (+3.03) and median of 2
admissions. The average number of hospitalization days was 12.65 (+12.37)
with a median of 9.5. Altogether 978 laboratory tests were requested. Among
these, 272 (28%) were not performed or the result was not found. Most
diagnoses belonged to the group of predominantly antibodies deficiencies with
15 cases (19%), followed by congenital defects of phagocytes with 6 cases
(8%), other well-defined immunodeficiency syndromes with 4 cases (5%),
diseases of immune dysregulation and combined T-cell and B-cell
immunodeficiencies, with 2 cases (3%) each, and complement deficiencies
with 1 case (1%). The group “other” comprised nine cases (11%) and 3 cases
(4%) had diagnosis of secondary immunodeficiency. The comparative analysis
of the time from the first visit to the definition of the diagnosis between patients
with or without the fulfilling of first time evaluation form was not statistically
significant (p> 0.93). The antibiotic prophylaxis was given to 58 cases (32%),
the management was expectant in 61 cases (34%) and human gammaglobulin
(IVIG) was given to 14 patients (8%). Hematopoietic stem cell transplantation
was performed in three patients. Most patients maintained regular follow-up
(54%), four patients (2%) died and in 15% of cases there was no record of
either follow-up consultation or death. The latter group was considered as lost
to follow-up.
LISTA DE SIGLAS
AD – Herança autossômica dominante
ADA - Adenosina deaminase
ALPS - Síndrome Linfoproliferativa Autoimune
AR – Herança autossômica recessiva
ATM - Ataxia-Telangiectasia Mutado
BCG – Bacilo Galmette-Guèrin
BAFFr - Receptor de fator ativador de célula B
BRAGID – Grupo Brasileiro de Imunodeficiência
Btk -Tirosina cinase de Bruton
C1INH- Inibidor de C1-esterase
CASP - Caspase
CD – Cluster de diferenciação
CDC – Centro de Controle de Doenças
CH- Complemento hemolítico
CINCA - Síndrome Neurológica, Cutânea e Articular Crônica Infantil
CVID - Imunodeficiência Comum Variável
DTH – Hipersensibilidade do tipo tardio (Delayed Type Hypersensitivity)
EAS – Exame de urina (uroanálise)
EDA-ID - displasia ectodérmica hipoidrótica com imunodeficiência
ESID - Sociedade Européia de Imunodeficiências (European Society for
Immunodeficiencies)
FCAS - Síndrome autoinflamatória ao frio familiar
FCUS - Urticária ao frio familiar
FMF - Febre Familiar do Mediterrâneo
G-CSF - Fator estimulador de colônia de neutrófilos
HIDS - Hiperimunoglobulinemia D e síndrome de febre periódica
HIV – Vírus da Imunodeficiência Humana
HPN - Hemoglobinúria paroxística noturna
HPV- Vírus Papiloma Humano
ICOS – Molécula co-estimulatória induzível
IDP – Imunodeficiência Primária
IFF – Instituto Fernandes Figueira
IFN - Interferon
Ig- Imunoglobulina
IKBA - Potenciador do inibidor de cadeia leve kappa em células B
IL- Interleucina
IPPMG – Instituto de Puericultura e Pediatria Martagão Gesteira
IRAK4 - Cinase associada ao receptor IL-1
IVIG – Imunoglobulina intravenosa
JAK- Cinase da família Janus
LAD – Deficiência de adesão leucocitária
LAGID – Grupo Latino Americano Para o Estudo das Imunodeficiências
Primárias (Latin American Group for the Study of Primary Immune Deficiencies)
LES- Lúpus Eritematoso Sistêmico
MAC - Complexo de ataque a membrana
MHC – Complexo principal de histocompatibilidade
MWS - Síndrome de Muckle-Wells
NBT- nitrobluetetrazolium
NEMO - Modulador essencial de NF-κB
NF-Κb - Fator nuclear κB
NK - Natural killer
NOMID -Doença Inflamatória Multisistêmica de Início Neonatal
PFAPA – Síndrome de Febre Periódica, Adenite Cervical, Faringite e Aftas
PPD – derivado proteico purificado
RAG - Gene ativador de recombinaseRNA –Ácido ribonucléico
RMRP- RNA de endoribonuclease processadora de RNA mitocondrial
SCID – Imunodeficiência Combinada Grave (Severe Combined Immune
Deficiency)
SHU - Síndrome hemolítico-urêmica
SUS – Sistema Único de Saúde
TACI- transmembrane activator and CAML interactor
TC – tomografia computadorizada
TCTH – Transplante de células-tronco hematopoiética
TLR - Receptor toll-like
TNF - Fator de necrose tumoral
TRAPS - Síndrome periódica associada ao receptor de fator de necrose
tumoral
VA- Via alternativa
VL - Via das lectinas
VP - Via clássica (classical pathway)
XL – herança ligada ao X
WASP - proteína da Síndrome de Wiskott-Aldrich
WHIM - síndrome de verrugas, hipogamaglobulinemia, infecções e
mielocatexia
SUMÁRIO
CAPÍTULO 1 - INTRODUÇÃO
14
CAPÍTULO 2 - REFERENCIAL TEÓRICO
16
2.1 . Conceito e historico
16
2.2. Epidemiologia
17
2.3. Classificação
19
2.4 Diagnostico
35
CAPÍTULO 3 - JUSTIFICATIVA
42
CAPÍTULO 4 - OBJETIVOS
45
CAPÍTULO 5 - MATERIAL E MÉTODOS
48
5.1. Desenho do estudo
48
5.2. Amostra
48
5.3. Local do estudo
48
5.4. Variáveis
49
5.5. Processamento dos dados
51
5.6. Análise dos dados
51
CAPITULO 6 - ASPECTOS ÉTICOS
52
CAPÍTULO 7 - RESULTADOS
53
7.1. Casuística
53
7.2 . Distribuição por gênero
53
7.3 . Distribuição por raça
55
7.4 . Origem
55
7.5 . Consangüinidade e história familiar
55
7.6 . Idade nos primeiros sintomas
55
7.7 . Idade na primeira visita
57
7.8. Motivo de encaminhamento e
origem de encaminhamento
57
7.9. Sítios de infecções
60
7.10 Sinais de alerta
64
7.11 Culturas
66
7.12 Internações
66
7.13 Hemotransfusões
69
7.14 Alterações do desenvolvimento psicomotor
69
7.15 Histórico de reações vacinais
69
7.16 Cicatriz de BCG
69
7.17 Histórico do parto
70
7.18 História alimentar
72
7.19 Hipóteses diagnósticas
72
7.20 Exames solicitados X exames realizados
75
7.21 Diagnóstico final
77
7.22 Meses entre primeiros sintomas e diagnóstico
79
7.23 Meses entre primeira consulta e diagnóstico
79
7.24 Análise comparativa do tempo decorrido até
o diagnóstico a partir do emprego ou não do
formulário de primeira vez
81
7.25 Seqüelas
85
7.26 Conduta terapêutica
85
7.27 Uso de gamaglobulina
87
7.28 Uso de antibiótico profilático
89
7.29 Uso de citocina terapêutica
91
7.30 Indicação de transplante de células-tronco
hematopoiéticas
91
7.31 Desfecho
91
CAPÍTULO 8 - DISCUSSÃO
92
CAPÍTULO 9 - CONCLUSÕES
105
CAPÍTULO 10 - REFERÊNCIAS
107
CAPÍTULO 11 - ANEXOS
112
CAPÍTULO 12 - APÊNDICES
134
CAPÍTULO 1 - INTRODUÇÃO
Crianças com infecções de repetição representam um problema de
saúde pública no Brasil. Em um país no qual, além da epidemia de Síndrome
de Imunodeficiência Adquirida (SIDA), ainda persistem diversos fatores
associados a causas secundárias de imunodeficiência, como desnutrição, más
condições de higiene, moradia e saneamento, o grupo de agravos a saúde
classificados como
imunodeficiências primárias ainda não conquistou seu
espaço entre as políticas de saúde públicas.
Entretanto, internacionalmente, o impacto que essas desordens
representam sobre o sistema de saúde já foi reconhecido (Lindegren et al,
2004; Espanõl, 2005). Com essa preocupação, países de todo o mundo já
organizaram políticas de saúde voltadas para o acompanhamento desses
pacientes
(Lindegren
et
al,
2004;
Zelazco
et
al,
1998;
http://www.imunopediatria.org).
No Rio de Janeiro, hospitais terciários como o Instituto de Pediatria e
Puericultura Martagão Gesteira
(IPPMG) ou o Instituto Fernandes Figueira
(IFF), com programas consolidados de treinamento em imunologia clínica,
ainda têm pouco a oferecer a essas famílias.
A proposta desta dissertação iniciou-se a partir da publicação de uma
portaria da Secretaria de Atenção a Saúde em dezembro de 2005,
estabelecendo o Instituto Fernandes Figueira como Centro de Referência em
imunodeficiências primárias, entre outras competências já consolidadas.
O objeto de estudo deste trabalho é a descrição do perfil clínico e
laboratorial dos pacientes que foram encaminhados para o serviço de alergia e
imunologia do Instituto de Pediatria Martagão Gesteira (IPPMG/UFRJ), entre
janeiro de 2006 e junho de 2008, com suspeita de imunodeficiência primária, e
de como está sendo realizado o acompanhamento e o diagnóstico desses
pacientes.
O ambulatório de alergia e imunologia do IPPMG é um serviço terciário
de referência no estado do Rio de Janeiro para o atendimento de pacientes
com imunodeficiência primária e, portanto, capaz de reunir pacientes com
condições ambientais e sócio-econômicas semelhantes, permitindo, dessa
forma, uma análise representativa do comportamento dessas desordens em
crianças brasileiras.
CAPÍTULO 2 - REFERENCIAL TEÓRICO
2.1. CONCEITO E HISTÓRICO
As imunodeficiências primárias (IDP) são desordens do sistema imune
caracterizadas por aumento da susceptibilidade à infecções, autoimunidade e
neoplasias malignas (Fischer, 2004; Stiehm et al, 2004). Defeitos genéticos e
hereditários em um ou mais componentes do sistema imunológico alteram o
funcionamento do mesmo, tornando o indivíduo vulnerável à infecções por
bactérias, vírus, fungos ou protozoários, com variados graus de gravidade
(Rosen et al, 1999).
Casos isolados de IDP foram relatados ao final da década de 20 (Stiehm
et al, 2004). Na década de 40, com o advento dos antibióticos tornando
possível a cura de infecções em indivíduos com o sistema imune preservado,
mais casos de imunodeficiências foram reconhecidos (Stiehm et al, 2004). Em
1952, Bruton descreveu um caso de um paciente com agamaglobulinemia que
se beneficiou do uso contínuo de gamaglobulina, mas somente na década de
80 a gamaglobulina venosa foi instituída como tratamento para as IDP (Stiehm
et al, 2004).
Nos últimos 50 anos, um enorme progresso ocorreu nesse campo, com
descobertas de novas imunodeficiências e instituição de tratamentos
promissores como transplante de medula óssea e terapia gênica (Good e
Verjee, 2001; Gaspar et al 2004). Os avanços da genética molecular e o
mapeamento do genoma humano permitiram a descrição de mais de 120 tipos
de IDP (Shearer et al, 2004; Geha et al, 2007;).
2.2. EPIDEMIOLOGIA
Os dados epidemiológicos sobre as IDP ainda são imprecisos devido à falta
de um sistema adequado de coleta de dados e registros na maior parte dos
países (Lindegren et al, 2004). A freqüência estimada é de 1:10.000 até
1:2.000 nascidos vivos (Bonilla e Geha, 2003; Bonilla et al, 2005). Na infância,
há uma predileção pelos meninos de 5:1, devido às síndromes ligadas ao X.
Quando o diagnóstico é feito em adultos a freqüência parece ser a mesma
em homens e mulheres (Knerr e Grimbacher, 2007).
Todos esses dados são, provavelmente, subestimados e imprecisos, em
função de, pelo menos, cinco fatores: a) falta de reconhecimento clínico, b)
falta de registro ou registro inadequado, c) superrepresentação de alguns
centros de referência, d) falta de padronização de definição de caso, e) morte
antes do diagnóstico (Lindegren et al, 2004).
Diversos países já desenvolveram seus sistemas de registros em nível
nacional, com objetivo de adquirir maior conhecimento sobre as IDP e melhorar
a qualidade do atendimento aos pacientes (Knerr e Grimbacher, 2007). No
Brasil existiram tentativas de implantar um sistema de registros próprio, já que
nosso país é um dos membros do LAGID (Latin American Group for the Study
of Primary Immune Deficiencies), grupo formado por 14 países, que possui um
sistema de registros já com 3321 pacientes (Knerr e Grimbacher, 2007).
Porém, esse número não reflete a prevalência atual, devido à capacidade
diagnóstica limitada dos países participantes e pelo fato dos dados terem sido
obtidos em algumas poucas cidades dos países envolvidos na pesquisa (Knerr
e Grimbacher, 2007).
A maioria dos países está implementando um sistema on line de registros,
semelhante ao utilizado pela Sociedade Européia para Imunodeficiências
(ESID-European Society for Immunodeficiencies) (Knerr e Grimbacher, 2007).
Em todos os países, as imunodeficiências predominantemente de
anticorpos aparecem como as formas mais freqüentes de IDP, com mais de
50% dos casos (Stiehm et al, 2004; Pérez et al, 2007; Knerr e Grimbacher,
2007).
Dados
do
ESID
de
2008
mostram
as
imunodeficiências
predominantemente de anticorpos com 55,25% dos casos, seguido de outras
imunodeficiências bem definidas (17,75%), deficiência de fagócitos (12,63%),
deficiências de células T (7,75%), defeitos do complemento (1,94%), doenças
de desregulação autoimune (1,15%) e doenças autoinflamatórias (1,08%)
(http://www.esid.org).
2.3. CLASSIFICAÇÃO
Por conta do acelerado reconhecimento de novos defeitos genéticos,
desde 1970, um comitê de especialistas se reúne a cada dois anos para
atualizar a classificação das IDP, baseada nos principais mecanismos
imunológicos implicados (Bonilla et al, 2005). A última atualização (anexo 3),
publicada em outubro de 2007 divide as imunodeficiências em oito grupos: a)
imunodeficiências combinadas T e B, b) deficiências predominantemente de
anticorpos, c) outras síndromes bem definidas, d) doenças de desregulação
imune, e) defeitos congênitos do número e/ou função de fagócitos, f) defeitos
na imunidade inata, g) doenças autoinflamatórias, h) deficiências de
complemento (Geha et al, 2007).
2.3.1. IMUNODEFICIÊNCIAS COMBINADAS DE CÉLULAS T E B.
Seus principais e mais graves representantes são as imunodeficiências
combinadas graves (SCID).
Pacientes com SCID apresentam grande comprometimento ou mesmo
completa ausência de imunidade específica, sendo susceptíveis à maioria dos
patógenos, inclusive patógenos oportunistas (Gaspar et al, 2001; Bonilla et al,
2005).
Os sintomas iniciais aparecem nos primeiros meses de vida com
infecções graves e recorrentes e desenvolvimento inadequado (Gaspar et al,
2001). Sem a reconstituição imunológica proporcionada pelo transplante de
medula, leva ao óbito na infância (Chan e Puck, 2005). Tratamentos
envolvendo terapia gênica estão em desenvolvimento e se mostram
promissores (Gaspar et al, 2004).
Diversas mutações genéticas já foram implicadas na patogenia das
diferentes formas de SCID (tabela 1).
Tabela 1: Mutações genéticas descritas na patogenia das SCID
Doença
SCID T- B+
Defeitos genéticos/patogenia
defeito de cadeia γ de receptores para IL-2, -4,
-7, -9, -15, -21
defeito em JAK 3
defeito na cadeia α de receptor de IL-7
defeito em CD 45
defeito de receptor de antígeno de célula T
SCID T- B- defeito completo de RAG 1 ou 2
defeito de proteína recombinase DNA-reparo
de Artemis
ausência de ADA, metabólitos linfotóxicos
elevados
maturação defeituosa de células T, B e células
mielóides
Herança
XL
AR
AR
AR
AR
AR
AR
AR
AR
Legenda:JAK cinase da família janus; RAG gene ativador de recombinase; ADA adenosina
deaminase.
Adaptado de Geha et al, 2007.
Mutações missense1 dos genes RAG 1, 2, Artemis, IL-7Rα ou RNA de
endorribonuclease
processadora
de
RNA
mitocondrial
(RMRP)
são
encontradas na Síndrome de Omenn, que cursa com diarréia protraída,
eritrodermia exsudativa, eosinofilia, adenopatia e hepatoesplenomegalia
(Roifman et al, 2006; Geha et al, 2007).
Deficiência de ZAP-70, MHC classe I e II, deficiência de canais de cálcio
entre outras, já foram descritas (Geha et al, 2007).
1.Mutação missense- mutação genética pontual, envolvendo a alteração de um único
nucleotídeo, podendo haver uma atividade residual do gene.
2.3.2. IMUNODEFICIÊNCIA PREDOMINANTEMENTE DE
ANTICORPOS
As imunodeficiências predominantemente de anticorpos (ou humorais)
formam um grupo bastante heterogêneo, com um espectro clínico que abrange
desde pacientes assintomáticos, até pacientes com graves manifestações de
imunodeficiências (Ballow, 2002). As doenças deste grupo resultam da
produção inadequada ou na função defeituosa de anticorpos devido à números
baixos de linfócitos B ou, ainda, da existência de células B com produção
inadequada de anticorpos (Lindegren et al, 2004).
Neste grupo de desordens, os pacientes apresentam infecções
sinopulmonares por bactérias encapsuladas como Streptococcus pneumoniae
ou Haemophilus influenzae tipo b (Ballow, 2002). Dependendo do tipo de
defeito, outros germes podem ser mais frequentemente envolvidos, como os
enterovírus, encontrados em pacientes com Agamaglobulinemia ligada ao X
(Webster, 1994; Ballow, 2002; Shiroma et al, 2004; Stiehm et al, 2004;) e
protozoários como a Giardia lamblia, em pacientes com deficiência seletiva de
IgA (Ballow, 2002). Os primeiros sintomas se iniciam normalmente após os
primeros seis meses a um ano de vida, quando ocorre a produção de
imunoglobulina endógena (IgG) em paralelo com a diminuição dos níveis de
imunoglobulina (IgG) maternos (Bonilla e Geha, 2003).
É o tipo mais comum de imunodeficiência primária, sendo responsável
por mais da metade dos casos registrados (Stiehm et al, 2004).
Mutações genéticas em moléculas envolvidas na diferenciação e
proliferação de células B estão sendo relacionadas como causas da
imunodeficiência primária sintomática mais comumente diagnosticada, a
imunodeficiência comum variável (CVID). Defeitos em cinco genes diferentes já
foram descritos: coestimulador induzível em células T(ICOS), TACI, CD19,
BAFF-r e MSH5; mas grande parte dos pacientes com CVID ainda não possui
defeito genético identificado ( Yong et al, 2008).
Pacientes com CVID apresentam infecções sinopulmonares de repetição
e
maior
tendência
a
manifestações
autoimunes,
neoplasias,
doença
granulomatosa e linfoproliferação (Cunningham-Rundles e Bodian, 1999). O
principal marcador diagnóstico é hipogamaglobulinemia (Weller e BankersFulbright, 2005).
A deficiência seletiva de IgA pode ser encontrada em indivíduos
saudáveis, assintomáticos. Porém, está associada a uma série de desordens
como: infecções sinopulmonares e gastrointestinais (giardíase de repetição),
autoimunidade e atopia (Cunninghan-Rundles, 2001). Alguns pacientes com
diagnóstico inicial de deficiência seletiva de IgA podem evoluir para CVID,
sugerindo uma causa comum para as duas doenças, pelo menos nesses casos
(Castigli e Geha, 2006).
A mutação no gene tirosina cinase de Bruton (Btk) é responsável pela
Agamaglobulinemia de Bruton ou deficiência de Btk, síndrome de herança
ligada ao X, em que o nível sérico de todos os isotipos de imunoglobulinas e
também o número de linfócitos B se encontram profundamente diminuídos
(Geha et al, 2007).
O tratamento dos defeitos humorais é baseado na administração regular
de gamaglobulina, por via intravenosa ou subcutânea, e antibioticoprofilaxia. O
uso da gamaglobulina aumenta a expectativa de vida e reduz a freqüência e a
gravidade das infecções (Wood et al, 2007).
2.3.3.
OUTRAS
SÍNDROMES
DE
IMUNODEFICIÊNCIA
BEM
DEFINIDAS
Este grupo passou a ser usado para englobar uma série de síndromes
bem caracterizadas clinicamente, nem todas com defeito gênico identificado, e
que não puderam ser enquadradas nos outros grupos já citados.
A síndrome de Wiskott-Aldrich é uma imunodeficiência ligada ao X cujo
fenótipo inclui trombocitopenia com plaquetas pequenas, eczema, infecções
recorrentes e maior incidência de manifestações autoimunes e malignidades
(Bonilla et al, 2005; Ochs e Thrasher, 2006). Ocorre por mutações do gene da
proteína da Síndrome de Wiskott-Aldrich (WASP). Otites médias recorrentes,
infecções sinopulomonares bacterianas e infecções virais são freqüentes,
assim como infecções oportunistas como pneumonia por Pneumocystis jiroveci
(Bonilla et al, 2005).
O sinal clínico mais característico é a trombocitopenia com plaquetas
pequenas. O paciente pode apresentar petéquias e sangramentos desde os
primeiros meses de vida (Ochs e Thrasher, 2006).
As respostas imunológicas dependentes de células B e T estão
afetadas, com diminuição de Imunoglobulinas G e M (IgA e IgE podem se
encontrar elevadas), defeito de resposta para antígenos polissacarídeos e
comprometimento progressivo de linfócitos T (Bonilla et al, 2005; Ochs e
Thresher, 2006). Atividade de células NK também se encontra diminuída
(Bonilla et al, 2005).
Ataxia-Telangiectasia é uma desordem decorrente de uma mutação no
gene Ataxia-Telangiectasia Mutado (ATM), localizado no cromossomo 11, de
herança autossômica recessiva, que cursa com alterações neurológicas
degenerativas, telangiectasias cutâneas e oculares, retardo de crescimento e
imunodeficiência (Nowak-Wegrzyn et al, 2004; Bonilla et al, 2005).
As síndromes de Hiper-IgE também fazem parte desse grupo, sendo
caracterizadas por IgE elevada no soro, dermatite e infecções recorrentes de
pele e pulmonares (Freeman e Holland, 2008). Pode ser autossômica
dominante (Síndrome de Job), por deficiência de STAT3, ou recessiva, por
mutação em TYK2 ou, ainda, por outras mutações ainda desconhecidas (Geha
et al, 2007).
São incluídas ainda neste grupo: anomalia de DiGeorge, displasias
imuno-ósseas,
candidíase
mucocutânea
crônica,
doença
veno-oclusiva
hepática com imunodeficiência e Síndrome de Hoyerall-Hreidarsson (Geha et
al, 2007).
2.3.4.DOENÇAS DE DESREGULAÇÃO IMUNE
O fenômeno de apoptose2 é importante para o equilíbrio da resposta
imune. Por conta dele, ocorre eliminação de células T autorreativas e limitação
de magnitude e duração de resposta à antigenos externos (Su e Lenardo,
2008). Defeitos em genes responsáveis pela regulação da apoptose de
linfócitos causam a Síndrome Linfoproliferativa Autoimune (ALPS)(Bleesing,
2002; Su e Lenardo, 2008).
A desregulação da homeostase dos linfócitos gera três conseqüências
principais: acúmulo anormal de linfócitos, autoimunidade pela falha de remoção
de linfócitos autorreativos e aumento na ocorrência de linfomas pela
sobrevivência inapropriada de linfócitos transformados (Bleesing, 2002). As
manifestações clínicas são: linfadenomegalias, esplenomegalia, citopenias
autoimunes, glomerulonefrite e linfomas (Bleesing, 2002). O achado laboratorial
característico é a presença de um número aumentado de linfócitos T (TCR
αβ+) duplo negativos (que não expressam CD4 ou CD8) no sangue periférico
(Bleesing, 2002).
A tabela 2 correlaciona a doença com o defeito genético.
2. Apoptose- morte celular programanda
Tabela 2: Defeitos genéticos descritos nas
Síndromes Linfoproliferativas Autoimunes (ALPS)
Doença
ALPS tipo 1a
Defeito de CD95(Fas)
Defeito genético
TNFRSF6
ALPS tipo 1b
Defeito de CD95l (FasL)
TNFSF6
ALPS tipo 2a
Defeito de caspase 10
CASP10
ALPS tipo 2b
Defeito de caspase 8
CASP8
ALPS N-Ras
Defeito de ativador de N-Ras
NRAS
Legenda:Casp- caspase; TNF – fator de necrose tumoral
Adaptado de Geha et al, 2007.
As síndromes de imunodeficiência com hipopigmentação também são
classificadas como doenças de desregulação imune. São decorrentes do
defeito no gene LYST, no caso da Síndrome de Chediak-Higashi, defeito de
RAB27A na Síndrome de Griscelli tipo 2 e defeito de AP3B1 na Síndrome de
Hermansky-Pudlak tipo 2 (Geha et al, 2007). Cursam com albinismo parcial e
baixa atividade de linfócitos T citotóxicos e células NK (Geha et al, 2007).
2.3.5. DEFEITO CONGÊNITOS DO NÚMERO E/ OU FUNÇÃO DE
FAGÓCITOS
Grupo caracterizado por apresentar infecções específicas para cada
defeito genético, como abscesso hepático na Doença Granulomatosa Crônica e
susceptibilidade à infecções por Mycobacterium sp. e Salmonella sp. nos
defeitos de interferon γ−interleucina 12 (IFNγ/IL-12/23). Esta doença não
costuma estar associada às infecções freqüentes nas outras formas de IDP,
particularmente
aquelas
vistas
nos
pacientes
com
imunodeficiência
predominantemente de anticorpos (Rosenzweig e Holland, 2004).
Defeitos no complexo enzimático responsável pela geração de
superóxido, necessário para auxiliar no processo de eliminação de bactérias
por fagocitose são a causa da Doença Granulomatosa Crônica (Rosenzweig e
Holland, 2004). O principal gene afetado é o gp91phox, de herança recessiva
ligada ao X, mas outros três genes autossômicos de herança recessiva podem
sofrer mutações, causando a doença (Rosenzweig e Holland, 2004; Seger,
2008).
Os principais germes isolados causadores de infecções nesses
pacientes
são:
Staphylococcus
aureus,
Burkholderia
cepacia,
Serratia
marcescens, Nocardia sp., and Aspergillus sp.; e os sítios de infecção mais
frequentes envolvem pulmões, pele, linfonodo e fígado (Rosenzweig e Holland,
2004). Ocorre formação de granulomas, abscessos de pele, abscesso
hepático, BCGíte, processo de cicatrização ruim, com eventual formação de
fístulas e pneumonia por fungos (Seger, 2008).
Deficiências de adesão leucocitária foram descritas e estão resumidas
na tabela 3.
Tabela 3: Deficiências de Adesão Leucocitária
Doença
Células
afetadas
Características clínicas
LAD 1
N+M+L+NK
retardo na queda do cordão umbilical, úlceras de pele,
periodontite e leucocitose
LAD 2
N+M
retardo na queda do cordão umbilical, úlceras de pele,
periodontite e leucocitose, grupo sanguíneo hh e retardo
mental
LAD 3
L+NK
retardo na queda do cordão umbilical, úlceras de pele,
periodontite e leucocitose com tendência a sangramento
Deficiência
de Rac 2
N
leucocitose e má cicatrização de feridas
Legenda:N- neutrófilos, M- monócitos e macrófagos, L- linfócitos, NK- natural killer, LADdeficiência de adesão leucocitária. Adaptado de Geha et al, 2007.
As neutropenias também fazem parte desse grupo. Podem ser graves como
a síndrome de Kostmann, que costuma ser fatal nos primeiros meses de vida,
ou mais brandas como a neutropenia cíclica (Dror e Sung, 2004).
Infecções de repetição são as principais manifestações clínicas, além de
gengivite crônica na neutropenia cíclica (Dror e Sung, 2004). O tratamento com
fator estimulador de colônia de neutrófilos (G-CSF) e, em casos específicos, o
transplante de medula óssea, melhoram a qualidade de vida desses pacientes
(Dror e Sung, 2004).
2.3.6. DEFEITOS DA IMUNIDADE INATA
A identificação dos receptores da família Toll (toll-like receptors – TLR)
permitiu a associação de algumas desordens dermatológicas a esse grupo de
imunodeficiências primárias (Kang et al, 2006). Os receptores TLR e as
moléculas envolvidas na transdução dos sinais gerados pelo engajamento
destes receptores são importantes componentes da imunidade inata. É através
desses receptores que a imunidade inata controla com precisão a ativação da
resposta adaptativa ( Kang et al, 2006). A tabela 4 sumariza os tipos de
imunodeficiências classificadas como defeitos da imunidade inata com os
defeitos genéticos descritos e as principais manifestações clínicas.
Tabela 4: Defeitos de imunidade inata
Doença
EDA-ID
EDA-ID
Deficiência de IRAK4
WHIM
Epidermodisplasia
verruciforme
Encefalite pelo Herpes
simplex
Encefalite pelo Herpes
simplex
Defeito genético
mutações de
NEMO (modulador
de ativação de NFκb
mutação de IκBA
resultando em
ativação
inadequada de
NF−κB
mutação de IRAK4
(um componente
da via de
sinalização de
TLR)
displasia ectodérmica
hipoidrótica+ deficiência de
célula T + infecções
mutação de
CXCR4 (receptor
para CXCL12)
hipogamaglobulilnemia+níveis
de célula B reduzidos+
neutropenia+
verrugas/infecção pelo HPV
mutação de
EVER1 e EVER2
mutação de
UNC93B1
mutação de TLR3
Manifestações clínicas
displasia ectodérmica
hipoidrótica+ deficiência de
anticorpos específicos +
infecções
infecções piogênicas
infecções pelo HPV e câncer
de pele
meningite e encefalite pelo
herpes simplex 1
meningite e encefalite pelo
herpes simplex 1
Legenda:EDA- ID- displasia ectodermica hipoidrótica com imunodeficiência; NEMOmodulador essencial de NF−κB ; NF−κB- fator nuclear κB; IκBA- potenciador do
inibidor de cadeia leve kappa em células B; IRAK- cinase associada ao receptor IL-1;
TLR- receptor toll-like; WHIM – síndrome de verrugas, hipogamaglobulinemia,
infecções e mielocatexia.
HPV- vírus papiloma humano. Adaptado de Geha et al, 2007.
2.3.7. DESORDENS AUTOINFLAMATÓRIAS
Grupo de desordens que não cursa com infecções de repetição, mas com
processos inflamatórios espontâneos, sem que haja títulos aumentados de
auto-anticorpos para justificar uma doença autoimune. Nove síndromes
autoinflamatórias hereditárias já foram identificadas (Padeh e Berkun, 2007).
• Febre Familiar do Mediterrâneo (FMF)
• Síndrome periódica associada ao receptor de fator de necrose tumoral
(TRAPS)
• Hiperimunoglobulinemia D e síndrome de febre periódica (HIDS)
• Síndrome autoinflamatória ao frio familiar (FCAS)/ urticária ao frio
familiar (FCUS)
• Síndrome de Muckle-Wells (MWS)
• Doença Inflamatória Multisistêmica de Início Neonatal(NOMID)/Síndrome
Neurológica, Cutânea e Articular Crônica Infantil (CINCA)
• Síndrome Blau
• Síndrome PAPA( artrite estéril piogênica, pioderma gangrenoso e acne)
• Síndrome de Mageed (osteomielite multifocal recorrente crônica)
O principal sinal é a febre recorrente, exceto na Síndrome de MuckleWells, cujos sintomas são urticária, perda auditiva e amiloidose (Geha et al,
2007). A maioria dos pacientes apresenta mutações em genes da família
da pirina ou da superfamília do fator de necrose tumoral (TNF) (Padeh e
Berkun, 2007).
Sintomas da FMF incluem: episódios de febre alta, acompanhados de
peritonite, sinovite e pleurite. A dor abdominal pode confundir com apendicite
aguda e, às vezes, se associada a vômitos e diarréia.
Manifestações
cutâneas, mialgia e dor articular também podem estar presentes (Padeh e
Berkun, 2007).
A síndrome PFAPA, caracterizada por crises episódicas de febre alta,
estomatite aftosa, faringite e adenite cervical, ainda não faz parte da
classificação de Geha e colaboradores, 2007, mas é considerada uma doença
autoinflamatória (Padeh e Berkun, 2007).
2.3.8. DEFEITOS DO COMPLEMENTO
Os defeitos do sistema Complemento compreendem um conjunto de
deficiências bastante raras, associadas as doenças autoimunes como o Lúpus
Eritematoso Sistêmico (LES) (Wen et al, 2004). Infecções recorrentes por
Neisseria gonorrhoeae ou N. meningitidis devem levantar a suspeita de
defeitos do complemento, mas infecções piogênicas também podem ocorrer
(Wen et al, 2004).
A deficiência de inibidor de C1-esterase (C1INH), um regulador da
cascata do complemento, resulta no Angioedema Hereditário, doença de
herança autossômica dominante que se caracteriza por crises recorrentes de
edema subcutâneo e submucoso em qualquer parte da pele ou dos tratos
respiratório e gastrointestinal (Gompels et al, 2005). O edema é indolor,
circunscrito e não pruriginoso, sem associação com urticária. Surge
espontâneamente ou após traumatismos e involui espontâneamente em 48 a
72 horas (Wen et al, 2004 e Gospels et al, 2005).
Outras deficiências e as síndromes associadas estão listadas na tabela
5.
Tabela 5: Deficiências de complemento
Síndrome
Infecções
Desordens
reumáticas
Dano renal
HPN
Componentes
Via
Herança
C1q, C1r, C1s,
C4, C2
VC
Autossômica
C3
VC, VA,VL
Autossômica
C5, C6, C7, C8,
C9
fator H, fator I
Todas (MAC)
Autossômica
VA (inibidores)
Autossômica
Properdina
ligado ao X
MLB
VA
(estabilizador)
VL
Autossômica
CR3
C1, C2, C4
Receptor
VC
Autossômica
Autossômica
MBL
C3
VL
VC, VA,VL
Autossômica
Autossômica
fator H
VA (inibidor)
Autossômica
CD46
fator acelerador
de decaimento,
CD59
inibidor de VA
inibidor de VA
e MAC
Autossômica
mutação
somática do
crom X
Correlação
clínica
LES, infecções
recurrentes
bacterianas
glomerulonefrite
infecções
piogênicas graves
infecções por
Neisseria
infecções
piogênicas,
Glomerulonefrite
SHU
infecções por
Neisseria
infecções
piogênicas e
sepse em crianças
e neonatos, LES
LAD
LES
LES
glomerulonefrite
membranoprolife
rativa e infecções
SHU atípico,
glomerulonefrite
SHU atípico
hemólise e
trombose
Legenda: VA - via clássica, LES - lupus eritematoso sistêmico, VA - via alternativa, VL - via das
lectinas (MBL), MAC - complexo de ataque a membrana, LAD - defeito de adesão leucocitária,
SHU - síndrome hemolítico-urêmica, HPN – hemoglobinúria paroxística noturna. Adaptado de
Wen et al, 2004.
2.4. DIAGNÓSTICO
A abordagem do paciente com suspeita de imunodeficiência primária se
inicia com a obtenção de história clínica e exame físico (Ballow e O’Neil, 2003)
A suspeita de uma imunodeficiência deve ser levada em consideração
frente a um paciente com infecções de repetição (Bonilla e Geha, 2003; Bonilla
et al, 2005; Stiehm et al, 2004; Rosen et al, 1999). A Fundação Jeffrey Modell,
junto com a Cruz Vermelha Norte Americana desenvolveram dez critérios
(anexo 1) que indicam a investigação de IDP (http://www.jmfworld.org). Esses
critérios já foram adaptados para nosso meio e podem ser verificados na figura
1.
Figura 1: Sinais de Alerta para investigação de IDP
Os 10 Sinais de Alerta para Imunodeficiência Primária na Criança
adaptados para o nosso meio são:
1. Duas ou mais Pneumonias no último ano
2. Quatro ou mais novas Otites no último ano
3. Estomatites de repetição ou Monilíase por mais de dois meses
4. Abscessos de repetição ou ectima
5. Um episódio de infecção sistêmica grave (meningite, osteoartrite,
septicemia)
6. Infecções intestinais de repetição / diarréia crônica
7. Asma grave, Doença do colágeno ou Doença auto-imune
8. Efeito adverso ao BCG e/ou infecção por Micobactéria
9. Fenótipo clínico sugestivo de síndrome associada a Imunodeficiência
10. História familiar de imunodeficiência
Adaptado pela Sociedade Brasileira de Pediatria (SBP), Associação Brasileira de Alergia e
Imunopatologia (ASBAI) e Grupo Brasileiro de Estudos de Imunodeficiências Primárias
(BRAGID)
da
fundação
Jeffrey
Modell
e
Cruz
Vermelha
Americana.
(http://www.imunopediatria.org.br)
Algumas infecções são mais relacionadas com determinados tipos de
IDP, o que pode ajudar a guiar os testes iniciais (Bonilla e Geha, 2003). Os
defeitos de anticorpos cursam com infecções sinopulmonares causadas por
bactérias encapsuladas, giardíase de repetição e enteroviroses (Bonilla e
Geha, 2003; Verbski et al, 2006). Defeitos combinados de célula T e B podem
apresentar qualquer tipo de infecção, inclusive por patógenos oportunistas
como P. jiroveci, enquanto que, nos defeitos de complemento, o patógeno mais
isolado é a Neisseria sp. (Bonilla e Geha, 2003; Verbski et al, 2006). A tabela 6
sumariza
os
imunológicos.
principais
patógenos
associados
aos
principais
defeitos
Tabela 6: Patógenos associados aos principais defeitos imunológicos
Organismo
Defeitos de
anticorpos
Vírus
ENTEROVIRUS
Bactéria
S pneumoniae, H
influenzae, S
aureus,
Pseudomonas
aeruginosa,
Clostridium fetus,
N meninigitidis,
Mycoplasma
hominis,
Ureaplasma
ureolyticum
Micobactéria
Fungo
INCOMUM
NÃO
Protozoário
Giardia lamblia
Deficiência
combinada
(T e B)
VÁRIOS
Defeitos de
fagócitos
INCOMUM
INCOMUM
Mesmos do
defeito de
anticorpo +
Listeria
monocytogenes,
Salmonella
typhi, microbiota
entérica
S aureus,
microbiota
entérica, P
aeruginosa, S
typhi,
Nocardia
asteroides
Mesmos dos
defeito de
anticorpo,
especialmente
N meninigitidis
Micobactérias
ambientais,
incluindo BCG
Micobactérias
ambientais,
incluindo
BCG
NÃO
Candida
albicans,
Histoplasma
capsulatum,
Aspergillus
fumigatus,
Coccidioides
immitis
A fumigatus,
C albicans
NÃO
P jiroveci,
Toxoplasma
gondii
P jiroveci
NÃO
Defeitos de
complemento
Legenda:BCG- Bacilo de Calmette Guérin.Adaptado de Bonilla e Geha, 2003.
A idade de início dos sintomas também pode ajudar. Quanto mais cedo
surgem os sintomas, mais grave deve ser o defeito (Ballow e O’Neil, 2003). As
deficiências combinadas se desenvolvem nos primeiros meses de vida,
enquanto que a Imunodeficiência Comum Variável pode surgir na vida adulta.
A história familiar pode ser sugestiva, já que muitas IDP são de herança
autossômica recessiva ou ligada ao X. Histórias de aborto recorrente,
consangüinidade, óbitos na infância por infecções graves ou diagnósticos de
imunodeficiência em parentes devem ser valorizadas (Ballow e O’Neil, 2003).
O exame físico deve ser completo, com parâmetros de crescimento,
procura de linfonodos palpáveis, tonsilas, dismorfismos, lesões de pele como
candidíase, úlceras, petéquias, eczema, cicatriz de abscessos, impetigo, entre
outras (Ballow e O’Neil, 2003).
Em 1999, Conley e colaboradores publicaram critérios para o
diagnóstico das principais imunodeficiências primárias (figura 2). São critérios
divididos em três categorias: definitivo, provável e possível. Pacientes com
critérios de diagnóstico definitivo possuem 98% de probabilidade de ainda ter o
mesmo diagnóstico em 20 anos. São os pacientes com a alteração genética
documentada. Pacientes sem o diagnóstico genético, mas com os exames
laboratoriais e clínicos compatíveis com a síndrome, têm o diagnóstico provável
e 85% de chance de manter o diagnóstico em 20 anos. Sem todas as
características clínicas e laboratoriais compatíveis com a imunodeficiência, o
diagnóstico é considerado possível.
Figura 2: Síndromes com critérios diagnósticos estabelecidos
IMUNODEFICIÊNCIA COMUM VARIÁVEL
DEFICIÊNCIA DE IgA
IMUNODEFICIÊNCIA COMBINADA GRAVE
SÍNDROME DE DiGEORGE
DEFICIÊNCIA DE MHC CLASSE II
DEFEITOS DE ADESÃO LEUCOCITÁRIA
DOENÇA GRANULOMATOSA CRÔNICA
IMUNODEFICIÊNCIA COMBINADA GRAVE LIGADA AO X
AGAMAGLOBULINEMIA LIGADA AO X
SÍNDROME HIPER IgM LIGADA AO X
ATAXIA TELANGIECTASIA
SÍNDROME DE WISKOTT-ALDRICH
SÍNDROME LINFOPROLIFERATIVA LIGADA AO X
Conley et al, 1999
Os
testes
diagnósticos
de
triagem
dos
principais
tipos
de
imunodeficiência podem ser divididos em quatro grupos, a seguir (Folds e
Schmitz, 2003; Verbsky et al, 2006; Azar e Ballas, 2007):
Suspeita de defeito combinado ou de células T:
•
Leucograma com diferencial de leucócitos
•
Subtipos de linfócitos (citometria de fluxo)-CD3, CD4, CD8,
CD19, CD56
•
Sorologias para antígenos vacinais: tétano, difteria e
pneumococo ( se negativos, revacinar e checar novamente
em 3-4 semanas)
•
Imunoglobulinas (IgA, IgG, IgM, IgE)
•
Testes de linfoproliferação
•
Considerar testes de hipersensibilidade tardia (testes
intradérmicos para candidina, tricofitina, streptoquinase,
PPD)
Suspeita de defeitos humorais:
•
Sorologias para antígenos vacinais : tétano, difteria e
pneumococo ( se negativos, revacinar e checar novamente
em 3-4 semanas)
•
Imunoglobulinas (IgA, IgG, IgE, IgM)
•
Considerar teste do suor para excluir fibrose cística e
tomografia computadorizada de tórax e/ou seios da face
•
Avaliação de subtipos de células B por citometria de fluxoCD19, CD20, CD27
•
Subclasses de IgG
•
Ensaio de proliferação de células B
•
Avaliação de depuração muco-ciliar (excluir Síndrome dos
cílios imóveis)
Suspeita de defeitos de fagócitos:
•
Contagem global e diferencial de leucócitos
•
Teste de nitroblue tetrazolium (NBT) ou diidro-rodamina
(citomertria de fluxo)
•
Ensaio de fagocitose ou ensaio de quimiotaxia
Suspeita de defeitos de complemento:
• C3, C4
• CH50
• C1INH
Causas de imunodeficiência secundária como HIV, desnutrição e
neoplasias devem ser excluídas. Abaixo, a figura 3 lista os principais exames.
Figura 3: Testes de triagem iniciais para excluir causas secundárias de
imunodeficiência
• Leucócitos com diferencial
• Bioquímica (creatinina, uréia, provas de função hepática, glicemia)
• EAS
• Proteínas totais e frações
• Sorologia anti-HIV
• Exames de imagem (radiografia de tórax, tomografia computadorizada
de tórax, radiografia de seios da face)
• Culturas apropriadas, se indicado
Adaptado de Azar e Ballas, 2007
CAPÍTULO 3 - JUSTIFICATIVA
A descoberta de mais de 120 tipos de defeitos do sistema imune, devido
ao crescimento das pesquisas genéticas (Fleisher e Oliveira, 2004), chamou a
atenção de especialistas em todo o mundo, que passaram a considerar essas
desordens como um problema de saúde pública (Lindegren et al, 2004).
Mesmo raras, as IDP são potencialmente graves, levam a seqüelas debilitantes
e a uma maior propensão a neoplasias malignas, gerando um custo elevado de
tratamento e acompanhamento (Espanõl et al, 2005).
Essa preocupação fez com que diversos países se reunissem em grupos
de estudo com os objetivos de conhecer melhor os dados epidemiológicos
dessas desordens, difundir conhecimento e proporcionar uma melhor qualidade
de vida para esses pacientes, organizando centros especializados para
diagnóstico, tratamento e acompanhamento dos mesmos. O LAGID ( Latin
American Group for the study of Primary Immune Deficiencies) realizou sua
primeira reunião em 1993 contando com poucos países como Brasil, Chile,
Argentina e Colômbia e, já em 2006, contava com a participação de treze
países (http://www.imunopediatria.org.br ).
Em 2001, baseando-se na premissa de que “as características que
definem as imunodeficiências primárias as tornam candidatas para uma
abordagem de intervenção de saúde pública” (Lindegren et al, 2004), o Centro
de Controle de Doenças (CDC) organizou um encontro entre especialistas em
diversas áreas relacionadas à imunodeficiência primária e saúde pública. O
resultado foi publicado em 2004 com recomendações para aplicação de
estratégias de saúde pública para o melhor atendimento dos pacientes com
imunodeficiência primária.
No Brasil, as IDP representam um alto custo para o Sistema Único de
Saúde (SUS) devido ao grande número de internações, muitas vezes
prolongadas ou em unidades de cuidados intensivos; dificuldades diagnósticas
com utilização de exames solicitados sem critérios adequados; uso de
antibioticoterapia
de
largo
espectro
e/ou
por
tempos
prolongados;
complicações da própria doença; alto custo do tratamento e risco maior de
desenvolvimento de autoimunidade e neoplasias.
Durante minha pós-graduação em Alergia e Imunologia, no Instituto de
Pediatria e Puericultura Martagão Gesteira (IPPMG), pude vivenciar as
dificuldades
do
acompanhamento
de
pacientes
com
suspeita
de
imunodeficiência. Os pacientes chegam ao ambulatório tardiamente, já com
seqüelas de difícil recuperação, não têm acesso a todos os exames para o
diagnóstico e o tratamento é difícil e oneroso.
Diante desse quadro, em 2002, profissionais de saúde envolvidos com
esse tema organizaram o Grupo Brasileiro para o Estudo de Imunodeficiências
Primárias (BRAGID) com objetivos de ( http://www.imunopediatria.org.br ):
1. Educação
de
médicos
sobre
diagnóstico
e
tratamento
das
Imunodeficiências Primárias no Brasil
2. Desenvolvimento de uma rede de laboratório por todo o país
3. Estabelecimento de uma rede de comunicação entre os centros de
referências para diagnóstico e tratamento de Imunodeficiências
Primárias
Em 22 de dezembro de 2005, a Secretaria de Atenção à Saúde resolveu,
através da Portaria número 745, estabelecer o Instituto Fernandes Figueira
como Referência Nacional para o Ministério da Saúde na Área de Saúde da
Mulher, Criança e Adolescente
para o desenvolvimento de ações de
articulação e assessoria à rede nacional para o cuidado dos pacientes de
imunodeficiência primária.
O estudo do perfil clínico e laboratorial dos pacientes com suspeita de
imunodeficiência primária permitirá um melhor conhecimento sobre o
comportamento dessas desordens em uma população brasileira e poderá
contribuir com novos estudos epidemiológicos necessários para se alcançar os
objetivos almejados pelo centro de referência.
CAPÍTULO 4 - OBJETIVOS
4.1. GERAL:
Descrever o perfil clínico e laboratorial dos pacientes com suspeita de
imunodeficiência primária atendidos no ambulatório de alergia e imunologia
pediátrica do IPPMG.
4.2. ESPECÍFICOS:
4.2.1. Identificar e classificar os pacientes com diagnóstico de IDP e
verificar, sempre que aplicável:
a) Gênero
b) Raça
c) Origem
d) Consangüinidade
e) História familiar de IDP
f) A idade de manifestações dos primeiros sintomas
g) Idade na primeira visita
h) Motivo de encaminhamento
i) Quem está encaminhando
j) Quais as principais manifestações clínicas apresentadas pelos
pacientes
k) Principais patógenos identificados
l) Número de internações em enfermarias e/ou unidades fechadas
m) Histórico de hemotransfusões
n) Alterações de desenvolvimento psicomotor
o) Reações vacinais
p) Histórico do parto
q) História alimentar
r) Hipóteses diagnósticas
s) Quais os exames que estão sendo realizados para o diagnóstico
t) Quais os principais diagnósticos
u) O tempo decorrido entre os primeiros sintomas e o diagnóstico
v) O tempo decorrido entre a primeira visita e o diagnóstico
w) Principais seqüelas
x) Quais os tratamentos que estão sendo instituídos para os
pacientes
y) Desfecho do paciente
4.2.2. Verificar se o formulário de primeira vez utilizado no serviço acelera a
definição do diagnóstico.
4.2.3 Propor um novo formato de formulário para a coleta de dados na
consulta de primeira vez e no acompanhamento dos pacientes no serviço.
CAPÍTULO 5 - MATERIAL E MÉTODOS
5.1. DESENHO DO ESTUDO
Foi realizado um estudo do tipo transversal descritivo retrospectivo
através de revisão de prontuários e formulário de primeiro atendimento no
serviço.
5.2. AMOSTRA
Foram incluídas no estudo todas as crianças com diagnóstico e/ou
suspeita
de
imunodeficiência
Imunodeficiências
primária
Primárias do Serviço
atendidas
no
de Alergia
e
ambulatório
de
Imunologia do
IPPMG/UFRJ no período de Janeiro de 2006 a Junho de 2008. Os pacientes
que permanecem em acompanhamento tiveram a coleta de dados censurada a
partir de junho de 2008 ou a partir da data de última consulta.
Os criterios de exclusão foram pacientes cujos prontuários ou fichas de
primeira vez não puderam ser acessados.
5.3. LOCAL DO ESTUDO
O estudo foi realizado no ambulatório de Imunodeficiências Primárias do
Serviço de Alergia e Imunologia do Instituto de Pediatria e Puericultura
Martagão Gesteira (IPPMG), na Universidade Federal do Rio de Janeiro
(UFRJ).
O IPPMG/UFRJ é um hospital pediátrico universitário , terciário, serviço
de referência em pediatria no Rio de Janeiro, que realiza cerca de 48.000
atendimentos ambulatoriais/ano, com 1.500 admissões de novos pacientes/ano
(dados do serviço do Arquivo Médico do IPPMG). Possui ambulatórios de
diversas especialidades que atende crianças com desordens
de alta
complexidade. O Serviço de Alergia e Imunologia recebe semanalmente
pacientes novos, referendados de todo o estado do Rio de Janeiro, e
eventualmente de outros estados, para consulta e avaliação alergoimunológica,
além de acompanhar regularmente pacientes com diagnóstico comprovado de
imunodeficiência primária. Em média são vistos 10 pacientes por semana, em
2 turnos: segunda-feira (tarde) e quarta-feira (manhã), incluindo as avaliaçãoes
de primeira vez, as consultas de seguimento e as infusões de gamaglobulina
intravenosa.
Os pacientes com suspeita de imunodeficiência primária respondem a
um formulário de primeiro atendimento criado pelo serviço ( anexo 2 ).
5.4. VARIÁVEIS
As informações dos prontuários e dos formulários de primeira vez foram
obtidas através de um instrumento de coleta de dados preenchido pelo
pesquisador (apêndice 1).
Os dados e as variáveis foram as seguintes:
• Identificação do paciente
• Registro da instituição
• Data de nascimento
• Data da primeira consulta
• Data do início dos sintomas
• Gênero
• Raça
• Município ou estado de origem
• Motivo do encaminhamento ao serviço
• Origem do encaminhamento
• Consangüinidade
• História familliar de imunodeficiência, sendo considerada positiva
quando algum membro da família tiver o diagnóstico de imunodeficiência
primária ou apresentar infecções frequentes, tendo ou não levado a
óbito do mesmo
• Histórico de infecções com patógenos isolados
• História do parto e neonatal com peso de nascimento, comprimento,
perímetro cefálico, apgar e intercorrências
• Alterações de desenvolvimento neuropsicomotor
• Internações em enfermaria e/ou unidades fechadas com tempo médio de
internação
• Reações vacinais, incluindo reações à BCG e necessidade de
tratamento
• História de hemotransfusões e possíveis reações
• Diagnóstico de doença do refluxo gastroesofágico
• Principais exames solicitados
• Conduta terapêutica
• Acompanhamento do uso de gamaglobulina com dose prescrita,
intervalo de doses, regularidade do uso, reações adversas
• Acompanhamento do uso de antibiótico profilático com tipo de antibiótico
prescrito, regularidade do uso, reações adversas
• Hipóteses diagnósticas prováveis
• Diagnóstico final
• Destino do paciente. Podendo ter alta, óbito ou seguimento
5.5. PROCESSAMENTO DOS DADOS
Os dados foram codificados, e transferidos para um banco de dados
utilizando o programa Excel®3 e processados utilizando os programas Excel® e
Statistica®4.
5.6. ANÁLISE DOS DADOS
As variáveis foram descritas através de freqüências absolutas e
percentuais para dados qualitativos e, através de médias, desvio padrão e
medianas para dados quantitativos.
Para testar a hipótese que o formulário de primeira vez utilizado no
serviço acelera o processo de definição diagnóstica, foi utilizado o teste não
paramétrico de Mann Whitney. A amostra foi dividida em três grupos:
Grupo A- pacientes com prontuário revisado
Grupo B- pacientes com prontuário e formulário de primeira vez revisados
Grupo C – pacientes com formulário de primeira vez revisados
3. Excel é um programa da Microsoft Office para Windows. Foi utilizada a versão para Windows 2007
4. Statistica é um programa da Starsoft , Inc. Foi utilizada a versão 7.0
Os grupos A e B foram comparados em relação ao tempo em meses
entre a primeira visita e o diagnóstico. Foi consideredo significativamente
estatístico o resultado com valor de p < 0.05.
A exploração gráfica foi feita através dos programas Excel® e
Statistica®.
CAPÍTULO 6 - ASPECTOS ÉTICOS
A pesquisa foi feita em consonância com o estabelecido na Resolução
no. 196/96 do CONEP/ Ministério da Saúde e suas complementares e com o
Código de Ética Médica de 1988. O projeto de pesquisa foi aprovado (Projeto
26/06) pelo Comitê de Ética em Pesquisa do Instituto de Pediatria e
Puericultura Martagão Gesteira/ UFRJ.
CAPÍTULO 7 - RESULTADOS:
7.1. Casuística
Foram encontrados registros de atendimento de 206 pacientes com
suspeita de imunodeficiência primária no ambulatório de Alergia e Imunologia
no período de janeiro de 2006 a junho de 2008, nas agendas de marcação do
serviço, nos formulários de primeira vez preenchidos e no arquivo nosológico
do serviço. Destes, 25 não puderam ser analisados em tempo hábil por
estarem passando por um processo de digitalização em uma firma tercerizada
ou não terem sido encontrados, e foram excluídos.
Foram incluídos 181 pacientes atendidos e que estavam de acordo com
os critérios de inclusão estabelecidos.
7.2. Distribuição por gênero
Dos 181 pacientes incluídos, 81(44,8%) eram do gênero feminino e 100
(55,2%) masculino, com um predomínio do sexo masculino de 1,23/1 (figura 4).
Figura 4: Distribuição percentual de pacientes de acordo com o gênero.
7.3. Distribuição por raça
Dados sobre a raça não foram informados em 87% dos pacientes e,
dessa forma, não foram analisados.
7.4. Origem
A maioria dos pacientes residia no município do Rio de Janeiro,
contabilizando 101 casos (55,8%). Havia dois pacientes (1,1%) de fora do
estado do Rio de Janeiro e oito pacientes (4,4%) sem a informação sobre o
endereço de residência. Dos 70 pacientes (38,7%) de outros municípios dentro
do estado do Rio de Janeiro, o município de Duque de Caxias foi a principal
origem relatada, com 11casos (6%).
7.5. Consanguinidade e história familiar
Apenas cinco pacientes (3%) apresentavam história de consanguinidade
e, em 63 casos (35%), o dado não foi coletado. A história familiar de IPD foi
positiva em 50 casos(28%) e 18 casos (10%) não continham o dado.
7.6. Idade nos primeiros sintomas
A idade de apresentação dos sintomas variou do nascimento a 132
meses, com média de 21,8 + 30 meses e mediana de 8 meses.
A figura 5 demonstra o número de ocorrências pelas idades.
Número de ocorrências
0
12
24
36
48 60 72 84
Idade (meses)
96
108 120 132 NA
Figura 5: Distribuição de pacientes pela idade dos primeiros sintomas.
O eixo horizontal (abscissas) corresponde às idades em meses e o eixo vertical
(ordenadas) ao número de pacientes. NA- dado não analisado
7.7. Idade na primeira visita
A idade na primeira visita variou de 0,2 a 203,51 meses com média de
67,52 +49,43 e mediana de 59,21meses. A curva de idade na primeira visita
apresentou dois picos: o primeiro nos primeiros 60 meses de vida e o segundo,
um pouco menor, dos 72 aos 120 meses (figura 6).
A figura 7 divide os
pacientes por faixa etária mostrando a maioria dos pacientes com idade de
primeira visita nos primeiros 10 anos de vida.
7.8. Motivo de encaminhamento e origem de encaminhamento
A ocorrência de infecções freqüentes foi o motivo de encaminhamento
em 135 pacientes (75%) dos casos. Porém, foi impossível analisar se os
pacientes se enquadravam entre os sinais de alerta por falta de informações
suficientes.
O local que mais encaminhou pacientes foi o IPPMG, com 60 pacientes
(33%) encaminhados. Os ambulatórios de Pediatria Geral e Hematologia foram
os principais serviços que encaminharam com 12 pacientes (6,6%) e 11
pacientes (6%) respectivamente. Em 94 casos (52%), os dados relativos a
origem do encaminhamento não puderam ser obtidos. Serviços da rede SUS
encaminharam 14 pacientes (8%).
Número de pacientes
Idade (meses)
Figura 6: Distribuição de pacientes pela idade na primeira visita. O eixo
das ordenadas corresponde ao número de pacientes e o eixo das abscissas
corresponde à idade em meses. Estão destacados pelas linhas em laranja os
picos de idade de encaminhamento ao Serviço de Referência.
Número de pacientes
Faixa etária (meses)
Figura 7: Distribuição de pacientes pela idade na primeira visita
separados por faixa etária. O eixo das ordenadas corresponde ao número de
pacientes e os valores dispostos abaixo de cada coluna correspondem à faixa
etária.
7.9. Sítios de infecções
A principal infecção encontrada foi Pneumonia5, com 119 casos(66%),
totalizando 18% do total de infecções. Noventa e dois pacientes (51%)
apresentaram diarréia, 80 pacientes (44%) apresentaram amigdalite, 66
pacientes (36%) apresentaram sinusite, 61 pacientes (34%) apresentaram
estomatite,
58 pacientes (32%) apresentaram infecções cutâneas, 53
pacientes (29%) apresentaram candidíase oral e/ou perineal/genital, 50
pacientes (28%) apresentaram otite média, 17 pacientes (9%) apresentaram
sepse, 11 pacientes (6%) apresentaram meningite, 3 pacientes (2%)
apresentaram osteomielite (tabela 7 )
A figura 8 mostra a distribuição dos sítios de infecções pelo número de
pacientes.
Em relação ao número de sítios de infecções por paciente, 7 pacientes
não apresentaram infecções e 1 paciente apresentou infecções em 9 sítios
diferentes. A figura 9 mostra a distribuição de sítios de infecções por pacientes.
Número de eventos infecciosos
Figura 8: Distribuição de número de eventos infecciosos por sítio.
Tabela 7: Distribuição absoluta e percentual de infecções por sítio
Infecções
Pneumonia
Total
119
%
66%
Diarréia
92
51%
Amigdalite
80
44%
Sinusite
66
36%
Estomatite
61
34%
Outros
60
33%
Infecções cutâneas
58
32%
Candidíase
53
29%
Otite média
50
28%
Sepse
17
9%
Meningite
11
6%
Osteomielite
3
2%
Número de pacientes
Figura 9: Distribuição de sítios de infecção por paciente. O eixo vertical
corresponde ao número de pacientes e o eixo horizontal ao número de sítios de
infecção.
7.10. Sinais de alerta
Os sinais de alerta referentes ao número de infecções apresentadas no
último ano não puderam ser analisadas porque os dados coletados não foram
considerados satisfatórios para análise.
Os sinais de alerta encontrados podem ser verificados na tabela 8. Asma
grave
e
doenças
autoimunes
estavam
presentes
em
4
pacientes
respectivamente, efeito adverso ao BCG foi relatado por 3 pacientes, fenótipo
sugestivo de síndrome associada a imunodeficiência foi visto em 5 pacientes,
infecção por micobactéria em 2 pacientes. Juntos, somam 10% dos casos. Pelo
menos 1 episódio de infecção grave foi visto em 28 pacientes (15,5%).
Tabela 8: Distribuição absoluta e percentual dos sinais de alerta
encontrados
No de
pacientes
%
Asma grave
4
0.02%
Doença autoimune
4
0.02%
Efeito adverso ao BCG
3
0.02%
Fenótipo sugestivo de síndrome associada a
imunodeficiência
5
0.03%
Infecção por micobactéria
2
0.01%
1 episódio de infecção grave (sepse, meningite,
osteoartrite)
28
15,5%
Sinais de alerta
7.11. Culturas
Apenas 26 resultados de exames microbiológicos foram encontrados. Os
resultados podem ser vistos na tabela 9.
7.12. Internações
Dos 181 casos, 122 (67%) relataram internações. Em 21% dos casos
não havia informações sobre o número de internações e, em 68% dos casos
não havia relato da quantidade de dias internado.
Em relação à unidades fechadas (UTI), 34 casos (19%)
relataram
internação. Em 2% dos casos não havia informação sobre o número de
internações e, em 35% dos casos não havia informação sobre a quantidade de
dias internado.
Em 15% e 41% dos casos não havia informação alguma referente à
internações e internações em unidades fechadas, respectivamente.
Os resultados de média, desvio padrão e mediana de internações em
enfermaria e unidades fechadas podem ser vistos na tabela 10 e os resultados
de média, desvio padrão e mediana de dias internados em enfermaria e
unidade fechada podem ser vistos na tabela 11.
Tabela 9: Patógenos identificados
Patógeno Identificado
Acinetobacter
Total
2
Bacilo de Koch
2
Campylobacter jejuni
1
Candida Albicans
1
E. Coli
1
Giardia lamblia
2
Herpes simples
2
Micobacteria atípica
1
Neisseria meningitidis
1
Proteus mirabilis
3
Pseudomonas aeruginosa
1
Rotavirus
1
Salmonella sp
2
Serratia
1
Staphylococcus aureus
1
Staphylococcus coagulase -
2
Staphylococcus α hemolítico
2
Total
26
Tabela 10: Número de internações (média, mediana e desvio
padrão) em enfermaria ou unidade fechada por paciente.
Internações
Enfermaria
UTI
Média (DP)
Mediana
3.25 (+3.03)
2
1.34 (+0.70)
1
UTI – unidade de tratamento intensivo, DP- desvio padrão
Tabela 11: Duração (média, mediana e desvio padrão) em dias das
internações em enfermaria ou unidade fechada
Internações
Enfermaria
UTI
média (+DP)
Dias internados
12.65 (+12.37)
mediana
9.5
19.81 (+15.47)
15
UTI – unidade de tratamento intensivo, DP- desvio padrão
7.13. Hemotransfusões
Havia relato de hemotransfusões em 26 casos (14%). Destes, 2
apresentaram reações. Não havia informação a respeito de reação em 4 casos.
Não havia relato de hemotransfusões em 26 casos (14%)
e o restante (71%)
negou hemotransfusão.
7.14. Alterações de desenvolvimento psicomotor
Havia relato de desenvolvimento psicomotor alterado em 32 casos
(18%). Não havia essa informação em 11 casos (6%).
7.15. Histórico de reações vacinais
Houve relato de reações vacinais em 13 pacientes (7%). Em 10
pacientes a reação foi à vacina de BCG e, destes, 5 não necessitaram de
tratamento e 1 dado não foi encontrado.
7.16. Cicatriz de BCG
Dos 181 pacientes, não havia relato sobre cicatriz de BCG em 171 casos
(94%). Três casos (2%) não apresentavam cicatriz e 7 casos (4%)
apresentavam.
7.17. Histórico do parto
Os pacientes foram divididos em três grupos de acordo com peso de
nascimento.
• Grupo de pacientes cujos dados não foram obtidos
• Grupo pacientes com peso de nascimento < 2,5 Kg
• Grupo de pacientes com peso de nascimento > 2,5 Kg
A maioria dos pacientes (73%) apresentava peso normal de nascimento,
acima de 2,5 Kg. Os resultados se encontram na tabela 12.
As médias, desvios padrões e medianas referentes ao peso de
nascimento, comprimento de nascimento e perímetro cefálico se encontram na
tabela 13.
A média de dias de queda do coto umbilical foi de 8,24 (+ 4,6) e mediana
de 7 dias. O número de dias variou de 3 a 30 dias. Em 37 (20,4%) casos não
havia essa informação.
As medidas de Apgar não foram obtidas em 123 casos 68%), sendo
insuficiente para análise.
O parto ocorreu sem intercorrências em 109 casos (60%) e 21 casos
(12%) não apresentavam esse dado.
Tabela 12: Distribuição de pacientes pelo peso de nascimento
Peso (Kg)
Pacientes
%
<2,500
22
12
>2,500
133
73
Dado não obtido
26
14
Tabela 13: Dados antropométricos (médias, medianas e desvio
padrão) mensurados ao nascimento
Dados de nascimento
Peso (Kg)
Comprimento (cm)
Perímetro cefálico (cm)
Média (+ DP)
Mediana
3,133 (+ 0,576)
3.23
48,9 (+ 3,1)
49
33,91 (+ 2,24)
34
Kg- quilograma; cm- centímetros; DP- desvio padrão
7.18. História alimentar
Os dados obtidos sobre a história alimentar e os resultados estão
resumidos na tabela 14.
7.19. Hipóteses diagnósticas
As hipóteses diagnósticas foram agrupadas de acordo com a
classificação de IDP publicada por Geha et al 2007 (anexo 3).
As imunodeficiências predominantemente de anticorpos foram as
principais hipóteses encontradas, com 47 casos (21%), seguidas por defeitos
congênitos de fagócitos com 32 casos (15%), outras síndromes de
imunodeficiência
bem
definidas
com
17
casos
(8%),
desordens
autoinflamatórias com 15 casos (7%), imunodeficiências combinadas com 10
casos (5%), doenças de desregulação imune com 7 casos (3%), defeitos do
complemento com 3 casos (1%) e defeitos de imunidade inata com 2 casos
(1%). Hipótese diagnóstica de afecção não pertencente ao grupo das IDP (não
imunodeficiência) foi encontrada em 27 casos (12%) e outras hipóteses
diagnósticas bem definidas em 11 casos (5%). Em 3 casos (1%) foram
encontradas hipóteses de imunodeficiências secundárias e em 46 casos (21%),
esse dado não foi obtido.
A figura 10 ilustra os resultados acima citados.
Tabela 14: distribuição de pacientes pela história alimentar
(aleitamento materno exclusivo por 6 meses, reação à proteína
heteróloga e refluxo gastroesofágico
Dados
Sim (%)
Não (%)
ND (%)
43 (24)
115 (64)
23 (13)
Reação à ptn heteróloga
18 (10)
141 (78)
22 (12)
RGE*
16 (9)
104 (57)
60 (33)
AME
* Um paciente apresentou o dado de RGE possível; AMEaleitamento materno exclusivo; RGE- refluxo gastroesofágico
ND- não divulgado
Figura 10: Distribuição percentual de hipóteses diagnósticas dos
pacientes encaminhados ao serviço, no momento da chegada, por grupo
de Imunodeficiência
7.20. Exames solicitados X exames realizados
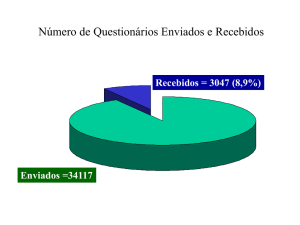
Ao todo foram solicitados 978 exames. Destes 272 (28%) não foram
realizados ou o resultado não foi encontrado. A tabela 15 relaciona os exames
e a freqüência com que foram solicitados e realizados.
Os hemogramas, imunoglobulinas, autoanticorpos, teste de Coombs,
sorologia anti-HIV, e teste cutâneo de leitura imediata (puntura) foram
realizados em mais de 80% dos casos. Testes intradérmicos para avaliação de
hipersensibilidade tardia (DTH), teste do suor, tomografia computadorizada
(TC) de tórax, foram realizados em 70 a 75% dos casos. Subclasses de IgG,
Diidro-rodamina (DHR), imunofenotipagem (contagem de células T, B e NK),
avaliação do complemento (C3, C4 e complemento hemolítico total), pesquisa
de deficiência de G-6PD, dosagem de iso-hemaglutininas, mielograma ou
biópsia de medula óssea, radiografia de tórax e radiografia de cavum foram
realizados em menos de 70% dos casos. A avaliação da resposta imune contra
antígenos polissacarídicos através da dosagem de anticorpos específicos após
vacina anti-pneumocóccica não-conjugada (23-valente) não foi realizada em
mais da metade dos casos em que foram solicitadas (59%).
Não foi encontrado relato de solicitação de teste de imunoproliferação.
Tabela 15: Ocorrências de exames solicitados e realizados por tipo de
exame
Exames
Solicitados
Realizados (%)
Hemograma
178
147 (83%)
Imunoglobulinas
179
146 (82%)
Sub-classes de IgG
49
25 (51%)
DHR
37
25 (68%)
Imunofenotipagem
93
48 (52%)
Avaliação de complemtento
104
66 (63%)
G-6PD
21
12 (57%)
Autoanticorpos
14
12 (86%)
Iso-hemaglutininas
25
17 (68%)
Mielograma/BMO
8
5 (63%)
Resposta à vacina antipneumocóccica
22
9 (41%)
Teste de Coombs
3
3 (100%)
Teste de linfoproliferação
0
0
Teste do suor
32
24 (75%)
Teste ID
33
23 (70%)
Sorologia anti-HIV
73
67 (92%)
Teste de puntura
23
20 (87%)
Radiografia de tórax
38
25 (66%)
TC de tórax
20
14 (70%)
Radiografia de cavum
26
17 (65%)
IgG- Imunoglobulina G; DHR- diidrorodamina; G6PD- glicose 6 fosfato
desidrogenase;
BMO- biópsia de medula óssea; ID- intradérmico; HIV- vírus da
imunodeficiência humana;
TC- tomografia computadorizada
7.21.
Diagnóstico final
O diagnóstico foi definido em 78 (43,1%) casos, sendo 36 (46%) não
imunodeficiências. Os diagnósticos foram distribuídos conforme o grupo da
classificação publicada por Geha et al em 2007.
A maioria dos diagnósticos pertenceu ao grupo de imunodeficiências
predominantemente de anticorpos com 15 casos (19%), seguido de defeitos
congênitos de fagócitos com 6 casos (8%), outras síndromes bem definidas
com 4 casos (5%), doenças de desregulação imune e imunodeficiências
combinadas, ambas com 2 casos (3%) e deficiências de complemento com 1
caso (1%) . O grupo outros totalizou 9 casos (11%) e 3 casos (4%) tiveram
diagnóstico de imunodeficiência secundária.
A figura 11 ilustra os percentuais de diagnósticos de imunodeficiência,
excluindo os casos de não imunodeficiência e imunodeficiência secundária.
Figura 11: distribuição percentual de diagnósticos definidos excluindo
casos de não imunodeficiência e imunodeficiência secundária
7.22. Meses entre primeiros sintomas e diagnóstico
A média de meses entre os primeiros sintomas e o diagnóstico foi de 41,17
(+40,74), variando de 0,43 a 192,17 meses, com mediana de 27,33.
7.23. Meses entre primeira consulta e diagnóstico
Para análise dessa variavél a amostra foi dividida em três grupos:
• Grupo A – pacientes com prontuário revisado
• Grupo B – pacientes com prontuário e formulário de primeira vez
preenchido
• Grupo C- pacientes com formulário de primeira vez preenchido
No grupo A o tempo variou de 5,98 antes de chegar ao ambulatório
(pacientes que já foram encaminhados com diagnóstico) a 112,31 meses, com
média de 15,04 (+ 33,25) e mediana de 4,47.
No grupo B o tempo variou de 7,5 meses antes de chegar ao ambulatório a
78,02 meses com média de 8,31 (+ 14,92) e mediana de 3,72.
No grupo C o tempo variou de 0 a 16,87 meses, com média de 4,98 (+
6,65) e mediana de 0,46.
No total o tempo variou de 7,5 meses antes de chegar ao ambulatório a
112,31 meses, com média de 9,22 (+ 19,11) e mediana de 3,72 (tabela 16).
Tabela 16: Duração em meses (média, mediana, desvio padrão,
mínimo e máximo) entre primeira consulta e diagnóstico
Grupos
Média (DP)
Mínimo
Máximo
Mediana
Grupo A
15,04 (+33,25)
(5,98)
11,.31
4,47
Grupo B
8,31 (+14,92)
(7,50)
78,02
3,72
Grupo C
4,98 (+6,65)
0
16,87
0,46
Total
9,22 (+19,11)
(7,50)
112,31
3,72
7.24. Análise comparativa do tempo decorrido até o diagnóstico à
partir do emprego ou não do formulário de primeira vez
Para saber se o formulário de primeira vez ajuda a acelerar o
diagnóstico foi utilizada a variável meses entre primeira consulta e diagnóstico
e comparados os grupos A e B. A fim de tornar a amostra mais homogênea
foram excluídos os casos com valores dois desvios padrão acima da média e
os pacientes que já chegaram ao ambulatório com diagnóstico. A tabela 17
demonstra os valores mínimos, máximos, médias, desvios padrão e medianas
dos grupos após a exclusão dos pacientes. O método utilizado para a análise
foi de Mann Whitney para variáveis não paramétricas e o resultado de p valor
foi de 0.93, não significativo.
Na figura 12 pode-se observar um gráfico em p-plot com os valores de
cada caso por grupo
No histograma apresentado na figura 13 podem ser observados os
números de observações pelos meses dos dois grupos sobrepostos.
Tabela 17: Duração em meses (mínimo, máximo, média, desvio padrão e
mediana) do tempo entre primeira consulta e diagnóstico, excluindo os
casos com valores dois desvios padrão acima da média e os pacientes
que já chegaram ao ambulatório com diagnóstico
Grupos
Grupo A
Grupo B
Mínimo
Máximo
Média (+ DP)
Mediana
0
22.26
6.57 ( + 7.97)
4.47
0
39.06
7.62 (+ 9.32)
4.14
2.5
2.0
Expected Normal Value
Desvio-padrão
1.5
1.0
0.5
0.0
-0.5
-1.0
-1.5
-5
0
5
10
15
20
25
30
35
40
45
Value
Tempo (meses)
Figura 12: Distribuição de casos pelo tempo em meses entre a primeira
consulta e o diagnóstico e o desvio padrão dos grupos A e B. Os pontos
em branco são referentes aos casos do grupo A e os pontos em vermelho são
referentes aos casos do grupo B. O eixo horizontal corresponde ao tempo em
meses e o eixo vertical ao desvio padrão.
11
10
9
Número de obsrevações
8
7
6
5
4
3
2
1
0
-5
0
5
10
15
20
25
30
35
40
Tempo (meses)
Figura 13: Distribuição de casos pelo tempo entre a primeira consulta e o
diagnóstico dos grupos A e B. O grupo A está representado pela cor
vermelha e o grupo B pela cor azul. O eixo vertical corresponde ao número de
observações e o eixo horizontal corresponde ao tempo em meses.
7.25. Seqüelas
Seqüelas foram relatadas em 14 casos (8%), sendo 11 pulmonares, 2
múltiplas e 1 neurológica.
7.26. Conduta terapêutica
A antibióticoterapia profilática foi indicada para 58 casos (32%), um
paciente (1%) fez tratamento com antibiótico, a conduta foi expectante em 61
casos (34%) e gamaglobulina humana (IVIG) foi indicada para 14 pacientes
(8%). Em 45 casos (25%) foram instituídas outras condutas e, em 24 casos
(13%), o dado não foi informado. (tabela 18).
Tabela 18: Distribuição absoluta e percentual por conduta terapêutica
Total
%
Antibioticoterapia profilática
58
32%
Antibioticoterapia tratamento
1
1%
IVIG
14
8%
Expectante
61
34%
Dado não obtido
24
13%
Outros
45
25%
Conduta terapêutica
7.27. Uso de gamaglobulina
Todos os pacientes com indicação de gamaglobulina iniciaram o
tratamento. Dos 14 pacientes em uso de IVIG, 8 pacientes (57%) tiveram o
tratamento suspenso por conta de desabastecimento da gamaglobulina
humana no SUS e 4 pacientes (29%) fizeram o tratamento regular. Não havia
o dado informado em 2 casos (14%) (tabela 19).
Foram relatadas reações adversas em 3 pacientes (21,4%), não havia
informação em 5 casos (35,7%) e 6 casos (42,9%) não apresentaram reações
(tabela 19).
Tabela 19: Distribuição absoluta e percentual de irregularidade do uso de
gamaglobulina e reações adversas
Uso de IVIG
Sim (%)
Não(%)
ND(%)
Irregularidade do tratamento
8 (57)
4(29)
2(14)
Reação adversa
3(21,4)
6(42,9)
IVIG- imunoglobulina venosa; ND- não divulgado
5(35,7)
7.28. Uso de antibiótico profilático
Dos 58 pacientes em uso de antibiótico profilático, usaram regularmente
31 pacientes (53%), o uso irregular foi relatado em 9 casos (16%) e 18 casos
(31%) não informaram. Apenas 1 relatou reação adversa. A tabela 20 mostra
os resultados relacionados ao uso de antibiótico profilático.
Tabela
20:
Distribuição
absoluta
e
percentual
de
antibioticoprofilaxia por tipo de antibiótico
Tipo de ATB
Total
%
27
47%
SMX-TMP
13
22%
SMX-TMP + amoxicilina*
6
10%
Associações
5
9%
Macrolídeo
1
2%
Outros
3
5%
Dado não obtido
3
5%
Amoxicilina
SMX-TMP- sulfametoxazol + trimetropim.
*SMX-TMP + amoxicilina- rodízio entre antibióticos
uso
de
7.29. Uso de citocina terapêutica
Apenas 2 pacientes tinham indicação de citocina terapêutica. Os 2 fizeram uso
regular de G-CSF e 1 paciente apresentou reação.
7.30. Indicação de Transplante de células-tronco hematopoiéticas
(TCTH)
A indicação de TCTH foi relatada em 10 pacientes. Três pacientes fizeram o
transplante fora do Rio de Janeiro. O TCTH realizado em 2 pacientes foi com
progenitores obtidos de sangue de cordão umbilical e placentário (SCUP). O
outro paciente submetido a TCTH não apresentou esse dado registrado em
prontuário.
7.31. Desfecho
A maioria dos pacientes manteve o seguimento, com 54% dos casos (97
pacientes). Tiveram alta 28 pacientes (15%),
e, em 28 casos, não foi
encontrado registro de consulta de seguimento ou óbito, sendo estes casos
caracterizados como abandono de acompanhamento (15%). Quatro pacientes
(2%) foram à óbito. O dado do desfecho não foi divulgado em 22 casos (12%)
e 2 pacientes (1%) foram encaminhados para outra especialidade por se
tratarem de casos de imunodeficiência secundária (HIV positivos).
CAPÍTULO 8 - DISCUSSÃO
Este trabalho apresenta um retrato dos aspectos clínicos e laboratoriais
de pacientes atendidos com suspeita de imunodeficiência primária em um
serviço de referência no Rio de Janeiro.
Outros trabalhos que apresentam aspectos semelhantes na literatura
são específicos para um tipo de diagnóstico (Kokron et al, 2004; CunninghamRundles e Bodian, 1999).
Rodrigues, em 1981, mostra um estudo retrospectivo de 11 pacientes
com suspeita de imunodeficiência primária e Grumach e colaboradores, em
1997 publicaram um artigo mostrando 15 anos de follow up de 166 casos de
imunodeficiência primária. A Fundação de Imunodeficiência Norte Americana
(Immune Deficiency Foundation) publicou, em 1995, um estudo observacional
sobre imunodeficiências primárias em todo o território norte-americano, mas
apenas para pacientes com o diagnóstico definido para imunodeficiência
primária (http://www.primaryimmune.org). Não foram encontrados artigos mais
recentes de perfil clínico de pacientes com suspeita de imunodeficiência
primária.
A fonte dos casos escolhida foi o Ambulatório de Imunodeficiência do
IPPMG, iniciado em janeiro de 2006 como uma extensão do ambulatório de
Alergia, que atendia e acompanhava pacientes com suspeita e/ou diagnóstico
de imunodeficiência primária. Com o aumento da demanda e a necessidade de
organizar melhor o serviço, foi criado um ambulatório específico para esses
casos, a partir de janeiro de 2006. O serviço atende pacientes de todo o estado
do Rio de Janeiro, como visto no estudo, e de outros estados do Brasil.
A coleta de dados foi censurada em junho de 2008 e, por isso, pacientes
ainda sem diagnóstico nesse período foram classificados como “sem
diagnóstico fechado”, pela data da última consulta.
Em relação a amostra estudada, a distribuição entre homens e mulheres
se mostrou bem semelhante, de 1,3:1, com um leve predomínio do gênero
masculino, como descrito por Stiehm e colaboradores, 2004; porém um pouco
menor do que o descrito por Knerr e Grimbacher, 2007, de 5:1.
A história familiar positiva para IDP foi de 28%, semelhante à encontrada
na literatura (Stiehm, 2004) e deve ser levada em consideração durante a
investigação (Lindegren et al, 2004). Consangüinidade dos pais não foi muito
freqüente na amostra estudada.
Em relação à idade dos primeiros sintomas, na amostra estudada foi
encontrada uma grande variação, do nascimento aos 132 meses. Stiehm, em
2004, refere que a idade dos primeiros sintomas varia de síndrome para
síndrome, o que pode ser a causa da variação encontrada no estudo. Apesar
da grande variação, a maioria dos pacientes da amostra iniciou os sintomas
nos primeiros meses de vida, conforme descrito por Stiehm, em 2004.
A idade na primeira visita apresentou dois picos, o primeiro nos
primeiros 60 meses de vida, quando é feito a maioria dos diagnósticos (Stiehm,
2004) e o segundo dos 72 aos 120 meses. Esse segundo pico, que vai até 10
anos de idade mostra um atraso no encaminhamento dos pacientes,
provavelmente por falta de conhecimento sobre as imunodeficiências (CostaCarvalho et al, 2005).
Vale ressaltar que o hospital é pediátrico e sua rotina permite a entrada
de pacientes novos até 12 anos e o acompanhamento de crianças até os 18
anos de idade, e as imunodeficiências primárias não podem mais ser
consideradas doenças apenas da infância (http://www.primaryimmune.org)).
A principal causa de encaminhamento encontrada na amostra estudada
foram as infecções de repetição. Segundo recomendações de Lindegren e
colaboradores, em 2004, o primeiro indício para o diagnóstico de IDP são as
infecções de repetição, persistentes, recorrentes e de difícil tratamento. Os
sítios de infecção encontrados seguiram o padrão descrito internacionalmente
(Ballow e O’Neil, 2003).
Em relação ao foco de infecção, foi impossível uma análise mais
detalhada dos casos do estudo, e uma correlação com os sinais de alerta já
descritos no capítulo de referencial teórico (http://www.imunopediatria.org). A
amostra estudada não continha dados sobre as infecções no último ano, com
foco, organismo identificado e resposta ao tratamento, como orientado por
Bonilla e colaboradores, em 2005.
A freqüência de cada sítio de infecção relatado pode ser analisada.
Pneumonia foi o principal sítio, com 66% dos casos. A sinusite, com 36% de
casos, foi observada com uma freqüência maior que a otite média aguda, com
28% dos casos. A sinusite não faz parte dos dez sinais de alerta adaptados
para o nosso meio (http://www.imunopediatria.org), mas está entre os sinais
originais, da Fundação Jeffrey Modell e a Cruz Vermelha Norte Americana
(anexo 1) (http://www.jmfworld.org).
Outras infecções vistas com freqüência e que não são associadas a
imunodeficiência primária são as infecções urinárias (no resultado do estudo
entrou como outras) e as amigdalites (44%), também relatadas por Kokron et
al, em 2004, nos casos de imunodeficiência comum variável.
Outros sinais de alerta como doença autoimune, fenótipo sugestivo de
imunodeficiência e efeito adverso ao BCG foram encontrados com uma
frequência baixa na amostra estudada (10%). Um episódio de infecção
sistêmica grave foi encontrado em 15,5% dos casos.
Em relação aos patógenos identificados, a amostra foi pequena, não
havendo registro suficiente para análise.
Em relação às internações, um número considerável de casos havia o
relato de internação (67%), com resultado semelhante ao encontrado pela
Fundação
de
Imunodeficiência
Norte
Americana,
que
foi
de
70%
(http://www.primaryimmune.org). A média de internações por caso foi de 3, 25
internações, um caso apresentou 15 internações. A média de dias internado
também foi alta (12,65 dias) comparando com a média de permanência em
internações hospitalares em pediatria do SUS no Rio de Janeiro no período de
janeiro de 2006 a dezembro de 2007, que foi de 9,4 (http://w3.datasus.gov.br).
As internações em unidades fechadas foram relatadas em 19% dos casos com
uma média de dias internado de 19,81 dias.
O custo dessas internações múltiplas para a saúde pública é enorme, o
valor médio de uma internação hospitalar em pediatria nos anos de 2006 e
2007 no município do Rio de janeiro foi de R$596,50 (http://w3.datasus.gov.br)
e o gasto total em internações em pediatria nesse mesmo período foi de
R$26.961.286,10. O impacto do diagnóstico precoce desses pacientes para os
recursos do SUS é significativo.
Em relação a hemotransfusões, alterações do desenvolvimento
psicomotor e reações vacinais a freqüência de pacientes com relato positivo foi
baixa. Vale ressaltar que, dos 13 pacientes que relataram reações vacinais, 10
(77%) foram relacionadas à BCG. Um paciente com agamaglobulinemia ligada
ao X apresentou quadro compatível com pólio associado ao vírus vacinal,
reação relatada em pacientes com defeitos de anticorpos (Ballow e O’Neil,
2003). Na amostra estudada apenas um paciente, com diagnóstico de
osteopetrose, referiu reação transfusional. As reações transfusionais estão
associadas a defeitos de célula B, por exemplo as reações anafiláticas em
pacientes com deficiência de IgA, ou defeitos combinados B e T nos quais
pode acontecer doença enxerto-contra-hospedeiro (Ballow e O’Neil, 2003).
Dados antropométricos e avaliação do grau de desnutrição não foram
suficientes para análise. O formulário de primeira vez possui um espaço para
peso e altura da criança, porém não são realizadas medidas sistemáticas e
anotadas em um gráfico. Um bom exame físico é essencial para iniciar uma
investigação diagnóstica (Ballow e O’Neil, 2003). Os dados antropométricos e
grau de nutrição não só são necessários para toda e qualquer criança que
esteja sendo avaliada, visto que algumas dessas crianças deixam de ir às
consultas de pediatria geral, como também fazem parte dos sinais de alerta já
citados anteriormente (anexo 1) (http://www.jmfworld.org). Além disso, a
desnutrição pode ser uma causa de imunodeficiência secundária, fazendo
parte do diagnóstico diferencial (Stiehm et al, 2004).
Quanto à presença de cicatriz de BCG, um dado de exame físico
importante para avaliar resposta à vacinação, quase a totalidade dos casos não
tinha registro desta informação (94%).
O histórico do parto não apresentou alterações significativas. A maioria
dos pacientes nasceu com peso normal (73%) e apenas 28% dos casos
apresentou alguma intercorrência neonatal. A queda do coto umbilical tardia (6
a 8 semanas) é um sinal visto em pacientes com defeitos de adesão de
leucócitos.( Ballow e O’Neil, 2003). Na amostra estudada a média informada de
dias de queda foi normal (8,24 dias).
Em relação as hipóteses diagnósticas, a maior freqüência foi de
deficiência de anticorpos (21%). Em segundo lugar ficaram os defeitos de
fagócitos (15%), seguido de outras síndromes bem definidas com 8%.
Entretanto, 56,9% de pacientes ainda não têm diagnóstico definido. As causas
da indefinição dos diagnósticos podem ser:
1. Pacientes novos no ambulatório, com apenas uma ou poucas
consultas no serviço
2. Testes diagnósticos indisponíveis no próprio Serviço ou em outra
unidade da rede do SUS
3. Dificuldade diagnóstica inerente às próprias doenças (IDPs)
Após a criação de um ambulatório específico para imunodeficiências, a
demanda aumentou e muitos pacientes iniciaram seu acompanhamento
recentemente. Desde o início do ano de 2008, 34(18,8%) pacientes iniciaram
acompanhamento e desde 2007, 99 (54,7%) pacientes procuraram o serviço.
Os testes de triagem mais simples como hemograma com diferencial de
leucócitos estão disponíveis facilmente, entretanto, exames considerados
essenciais para uma investigação inicial, muitas vezes não são encontrados no
SUS. Os testes de imunoglobulinas e imunofenotipagem de linfócitos são
extremamente irregulares, muitas vezes se encontrando em falta; subclasses
de imunoglobulinas e testes para anticorpos específicos contra pneumococos
não são disponíveis no Rio de Janeiro, exceto em laboratórios privados. O
teste de oxidação da diidrorodamina 123, para o diagnóstico de defeito de
fagócitos está sendo regularmente realizado pelo Laboratório de Fisiopatologia
Humana do IFF/Fiocruz desde o segundo semestre de 2007.
Outros exames de maior complexidade, como testes genéticos e
moleculares ainda não estão disponíveis no Brasil ou somente à titulo de
pesquisa em alguns centros. As causas de muitas imunodeficiências ainda não
foram descobertas e, portanto, não existem protocolos.
Na amostra estudada, foram solicitados 978 exames, dos quais 28% não
foram realizados ou não foram encontrados resultados em prontuário. Como
discutido acima, os exames necesários para diagnóstico muitas vezes não
estão disponíveis. Dessa forma, duas situações podem ocorrer: o médico
assistente pode não solicitar o exame que já sabe que não poderá ser feito,
como no caso dos testes de linfoproliferação; ou, quando o exame é feito de
maneira irregular, o médico assistente solicita e nem sempre é realizado como
é o caso da resposta à vacina antipneumocóccica, com apenas 41% dos testes
realizados, nem sempre nos prazos adequados ao diagnóstico. Dos 20 exames
analisados, só o teste de Coombs teve retorno de resultados em 100% dos
casos em que houve solicitação.
Lindegren e colaboradores, em 2004, sugerem ferramentas para o
desenvolvimento de políticas públicas de saúde. As ferramentas são:
• estudos observacionais para coleta, análise e interpretação de
dados relacionados às IDP;
• pesquisa epidemiológica com o estudo de distribuição das
doenças, relações entre fenótipo-genótipo e estudos que
contribuam com o conhecimento mais aprofundado do curso
clínico e história natural dessas desordens;
• ciência
e
capacitação
laboratorial
em
conjunto
com
as
ferramentas acima para diagnóstico, caracterização fenotípica,
elaboração de testes de triagem e testes diagnósticos.
A capacitação laboratorial da rede de Saúde Pública parece ser um dos
pontos principais para a melhoria da assistência à esses pacientes.
Dentre os diagnósticos definidos, excluindo os casos de não
imunodeficiência, a freqüência maior foi de defeitos de anticorpos, compatível
com a literatura. Dados do ESID (http://www.esid.org) demonstram uma
freqüência de 54,36% de pacientes com diagnóstico de deficiência de
anticorpos. Na amostra estudada a freqüência foi menor (39%). Em segundo
lugar, ainda de acordo com os dados do ESID, estão as outras
imunodeficiências bem definidas com 17,68%. Na amostra estudada esse
diagnóstico representou 10% dos casos, ficando em quarto lugar. Desordens
de fagócitos têm uma freqüência pelo ESID de 12,57% e na amostra estudada
a freqüência foi de 15%. As deficiências de célula T apresentam freqüência de
8,35% enquanto que, na amostra estudada, foi de 5%. Deficiências do
complemento apresentam freqüência de 2,07% no ESID e 3% na amostra.
Doenças de desrregulação imune apresentam freqüência de 1,31% no ESID e
5% na amostra. Doenças autoinflamatórias aparecem com 1,06% no ESID e
não aparecem na amostra, apesar de a amostra ter um diagnóstico de PFAPA,
que é considerado uma desordem autoinflamatória, mas ainda não está na
classificação mais recente disponível na literatura, sendo incluída como outros
diagnósticos. Outras imunodeficiências não classificadas apresenta freqüência
de 2,6% enquanto que na amostra estudada são responsáveis por 23% dos
casos.
Essas diferenças encontradas podem se dever ao fato de a amostra ser
pequena e do ambulatório acompanhar alguns pacientes com diagnósticos de
doenças tradicionalmente vistas por algumas outras especialidades, como o
serviço de Hematologia, que concentra casos de Osteopetrose, síndrome de
Wiskott-Aldrich e síndrome de Chediak-Higashi.
O tempo entre os primeiros sintomas e o diagnóstico, medido em meses,
apresentou uma grande variação, de 0,43 a 192,17 meses, com média de
41,17 e mediana de 27,33 meses. Essa variação provavelmente se deve ao
fato de que os pacientes podem ser encaminhados com muito atraso para o
serviço, passando antes por diversas especialidades e diversas internações
antes de chegarem ao ambulatório, gerando muitos custos desnecessários
para a Saúde Pública. O segundo pico de idade da primeira visita verificado na
figura 6 pode demonstrar esse atraso na chegada do paciente. Segundo
Stiehm, 2004, 40% dos diagnósticos são feitos no primeiro ano de vida e 40%
nos primeiros 5 anos de vida. A curva tem o seu segundo pico dos 6 aos 10
anos de vida.
Mesmo depois de encaminhados, esses pacientes ainda podem demorar
a fazer o diagnóstico como pode ser visto no tempo entre primeira visita e
diagnóstico. A variação foi de 0 (excluindo-se os pacientes que já chegaram
com diagnóstico) a 112,31 meses, com média de 9,22 e mediana de 3,72. A
diferença entre a média e a mediana e o desvio padrão alto encontrado devese ao fato da amostra ser bastante heterogênea.
O Ambulatório de Imunodeficiências Primárias utiliza um formulário de
primeira vez (anexo 2) para tentar padronizar uma coleta de dados dos
pacientes que acelere o processo de diagnóstico. Esse formulário, em conjunto
com o prontuário, foram as fontes de informação do presente estudo.
A análise estatística feita para verificar a hipótese de que o formulário de
primeira vez utilizado acelera o processo do diagnóstico não mostrou
significado estatístico, ou seja, não parece ajudar. A variável utilizada foi o
tempo entre a primeira visita e o diagnóstico em meses. Para tornar as
amostras mais homogêneas foram excluídos os resultados dois desvios
padrões acima da média e os pacientes que já iniciaram o acompanhamento
com o diagnóstico definido. Quando dispostas nas figuras 12 e 13
demonstrados no capítulo de resultados, as amostras analisadas se
sobrepõem, mostrando sua semelhança. As possíveis causas desse resultado
são:
1. amostra pequena, não tendo dados suficientes para um resultado
significativo;
2. o formulário de primeira vez não atende ao objetivo proposto, não há
campos para informações mais detalhadas sobre infecções no último
ano, internações, outros sinais de alerta associados, hipóteses
diagnósticas;
3. coleta de dados imprecisa pelo atendente no momento da coleta,
resultando em dados insuficientes para análise;
Dessa forma, o presente estudo se propõe a sugerir um novo formulário
de primeira vez, com campos mais detalhados para informações já presentes
no formulário antigo e campos novos para as perguntas não contidas
previamente. (apêndice 2) Também é sugerido um formulário de seguimento do
paciente para padronizar as consultas e melhorar a coleta de dados durante a
assistência (apêndice 2).
Outra variável dificilmente encontrada na revisão dos casos foi em
relação às seqüelas. A maioria não tinha essa informação. Foram encontradas
seqüelas em 14 (8%) pacientes, a maioria sendo pulmonares.
Em relação ao tratamento, o serviço tem como rotina iniciar
antibioticoprofilaxia para todos os pacientes com evidências fortes de
imunodeficiência e infecções de repetição antes do diagnóstico definitivo. Os
principais antibióticos prescritos são a amoxicilina e o sulfametoxazoltrimetropim. É de consenso geral que a profilaxia com antibiótico para
determinadas imunodeficiências é benéfica (Stiehm, 2004; Wood et al, 2007).
O uso de gamaglobulina humana (IVIG) foi indicado para 14 pacientes e
iniciado por todos eles. Entretanto, não houve regularidade no tratamento de 8
(57%) pacientes devido à falta do medicamento no SUS. A IVIG é o tratamento
de escolha para pacientes com defeitos de anticorpo e infecções de repetição.
Comprovadamente, a IVIG diminui o número e a gravidade das infecções,
melhorando a qualidade de vida e o prognóstico do paciente (Wood et al,
2007).
O uso de citocina terapêutica foi indicado para dois pacientes , um com
neutropenia crônica associada a glicogenose do tipo 1b e outro com
neutropenia cíclica, sem maiores intercorrências.
Em 10 casos houve indicação de transplante de células-tronco
hematopoiéticas (TCTH) e 5 realizaram o procedimento, dois pacientes foram à
óbito pós transplante e os outros 3 estão em seguimento e em bom estado de
saúde. Houve 1 caso de perda do seguimento e 3 óbitos antes do transplante.
Ainda em seguimento estão 2 pacientes aguardando transplante. Esses dados
diferem dos apresentados na seção de resultados pois refletem informações
recebidas das instituições nas quais os transplantes foram realizados, porém
não constam dos prontuários, que cessam de registrar as consultas a partir da
data de encaminhamento ao centro de transplante.
A freqüência de perda de seguimento foi de 15%, talvez um outro fator
que impeça mais diagnósticos. A demora no diagnóstico e o baixo poder
aquisitivo da amostra podem ser causas da taxa de abandono. Os dois
pacientes encaminhados para outro serviço tiveram diagnóstico sorológico de
infecção pelo HIV e encaminhados para o serviço de Infectologia do hospital.
Os quatro pacientes que foram à óbito, já referidos acima, dois tinham
diagnóstico de imunodeficiência combinada grave, um apresentava doença
granulomatosa crônica e o outro apresentava osteopetrose.
CAPÍTULO 9 - CONCLUSÕES
9.1. O estudo presente fez uma descrição do perfil clínico e laboratorial de
pacientes com suspeita ou diagnóstico de imunodeficiência primária
acompanhados em um serviço de referência de Rio de Janeiro e
encontrou um total de 39 pacientes com diagnóstico confirmado
dentro de um universo de 181 pacientes avaliados. Os diagnósticos
observados, a distribuição dos casos de acordo com o mecanismo
imunitário comprometido e os protocolos de investigação empregados
no serviço de referência foram semelhantes aos observados em
outras séries de pacientes descritas na literatura.
9.2. A inexistência de alguns exames complementares ou a irregularidade de
sua oferta na rede do SUS foi observada para a maioria dos pacientes
atendidos e teve impacto negativo sobre a agilidade do diagnóstico,
com consequente prejuízo na qualidade da assistência prestada.
9.3. O tratamento dos pacientes com diagnóstico firmado de IDP ou suspeita
de IDP foi conduzido de acordo de acordo com as diretrizes
internacionais e as especificidades de cada caso, e consistiu
majoritariamente de: antibioticoterapia profilática, imunoglobulina
intravenosa
e
hematopoiéticas.
indicação
de
transplante
de
células-tronco
9.4. O formulário de consulta de primeira vez em uso no Serviço de
Referência
(anexo
2)
não
se
mostrou
superior
ao
registro
convencional em prontuário médico em termos de acelerar o
diagnóstico dos casos encaminhados com suspeita de IDP, embora a
imprecisão no preenchimento ou a ausência de dados registrados
possa ter interferido na análise desta hipótese.
9.5. Os dados obtidos neste estudo permitem a proposição de um novo
instrumento de avaliação clínica, para registro de anamnese, exame
físico e resultados de exames complementares, tanto de consulta de
primeira vez quanto de consultas de seguimento (apêndice 2). É
recomendável que este novo instrumento seja validado em escala
piloto, como estratégia de aprimoramento.
CAPÍTULO 10 - REFERÊNCIAS
• Azar A, Ballas Z. Evaluation of the adult with suspected
immunodeficiency. The American Journal of Medicine 2007; 120:764768.
• Ballow M, O’Neil K. Approach to the Patient with Recurrent Infections. In:
Adkinson N, Yunginger J, Busse W, Bochner B, Holgate S, Simons E.
Middleton’s Allergy Principles and Practice 6a Ed.Philadelphia, Mosby,
2003.
• Ballow M. Primary immunodeficiency disorders: Antibody deficiency. J
Allergy Clin Immunol 2002; 109:581-91.
• Bleesing J. Autoimmune lymphoproliferative syndrome. A genetic
disorder of abnormal lymphocyte apoptosis. Immunol Allergy Clin N Am
2002; 22: 339-355.
• Bonilla FA, Bernstein L, Khan DA, Ballas ZK, Chinen J, Frank MM, et al.
Practice Parameter for the diagnosis and management of primary
immunodeficiency. Ann Allergy Asthma Immunol 2005; 94: S1-S61.
• Bonilla FA, Geha RS. Immunologic Disorders. Primary immunodeficiency
diseases. J Allergy Clin Immunol 2003; 111: S571-81.
• BRAGID. 10 Sinais de Alerta para Imunodeficiência Primária na Criança
adaptados para o nosso meio. http://www.imunopediatria.org. Sd.
• BRAGID. Objetivos. http://www.imunopediatria.org. Sd
• Bruton O. Agammaglobulinemia.Pediatrics 1952;9:722-728.
• Castigli E, Geha R. Molecular Basis of Commom Variable
Immunodeficiency. J Allergy Clin Immunol 2006; 117: 740-746.
• Chan K, Puck J. Development of population-based newborn screening
for severe combined immunodeficiency. J Allergy Clin Immunol 2005;
115:391-398
• Conley ME, Notarangelo LD, Etzioni A. Diagnostic Criteria for Primary
Immunodeficiencies. Clin Immunol 1999; 93:190-7.
• Costa-Carvalho B, Condinoneto A, Conley M, Moraespinto M,
Carneirosampaio M. The awareness of pediatricians about primary
immunodeficiencies in Brazil. In: Primary immunodeficiencies
Consortium Conference, 2005. Clin Immunol 2005; 116:285.
• Cunningham-Rundles C, Bodian C. Commom Variable
Immunodeficiency: clinical and immunological features of 248 patients.
Clin Immunol 1999; 92(1): 34-48.
• Cunningham-Rundles C. Physiology of IgA and IgA deficiency. J Clin
Immunol 2001; 21(5): 303-309.
• Dror Y, Sung L. Update on Child Neutropenia: Molecular and Clinical
Advances. Hematol Oncol Clin N Am 2004; 18: 1439-1458
• ESID. Distribuição dos principais grupos de imunodeficiência primária.
http://www.esid.org/statistics.php?sub=2 acessado em 08/08/08.
• Espanõl T, Hernández M, Giner MT, Casas C, Gurbindo D, Marco T, et
al. Directorio de pruebas diagnósticas de las inmunodeficiencias
primarias. Allergol et Immunopathol 2005; 33 (3):157-61.
• Fischer A. Human primary immunodeficiency diseases: a perspective.
Nat Immunol 2004; 5: 23-30.
• Fleisher T A, Oliveira JB. Functional and molecular evaluation of
lymphocytes. J Allergy Clin Immunol 2004; 114:227-34.
• Folds J, Schmitz JL. Assessment And Modulation Of The Immune
Response. Clinical and laboratory assessment of immunity. J Allergy Clin
Immunol 2003; 111:S702-11.
• Freeman A, Holland S. The Hyper-IgE Syndromes. Immunol Allergy Clin
N Am 2008; 28: 277-291.
• Gaspar H, Gilmour K, Jones A. Sevdere combined immunodeficiencymolecular pathogenesis and diagnosis. Arch Dis Child 2001; 84: 169173.
• Gaspar H, Parsley K, Howe S, King D, Gilmour K, Sinclair J et al. Gene
Therapy of X-linked Severe Combined Immunodeficiency By Use of a
Pseudotyped Gamma Retroviral Vector. Lancet 2004; 364:2181-2187.
• Geha R, Notarangelo L, Casanova J, Chapel H, Conley ME, Fischer A et
al. Primary immunodeficiency diseases: An update from the International
Union of Immunological Societies Primary Immunodeficiency Diseases
Classification Committee. J Allergy Clin Immunol 2007; 120: 776-94.
• Gompels M, Lock R, Abinun M, Bethune C, Davies G, Grattan C et al C1
inhibitor deficiency: consensus document. Clin Exp Immunol 2005; 119:
379-394.
• Good R, Verjec T. Historical and Current Perspectives on Bone Marrow
Transplantation for Prevention and Treatment of Immunodeficiencies and
Autoimmunities. Biol Blood Marrow Transplant 2001; 7; 123-135.
• Grumach A. J., Duarte A, Bellinati-Pires R, Pastorino A, Jacob C, Diogo
C et al. Brazilian Report on Primary Immunodeficiencies in Children: 166
cases studied over a follow up ttime of 15 years. Clin Immunol 1997;
17(4): 340-345.
• Informações em saúde: internações por especialidade e local de
internação de 1981 a 2007. DATASUS.
http://w3.datasus.gov.br/datasus/datasus.php?area=359A1B375C2D0E0
F359G902H0I1Jd2L22M0N&VInclude=../site/infsaude1.php&lista=op1
(acessado em 7/8/8).
• Jeffrey Modell Foundation. 10 Warning Signs of Primary
Immunodeficiency. http://www.jmfworld.org. sd.
• Kang S, Kauls L, Gaspari A. Toll-like receptors: Applications to
dermatologic disease. J Am Acad Dermatol 2006;54:951-83
• Knerr V, Grimbacher B. Primary Immunodeficiency registries. Curr Opin
Allergy Clin Immunol 2007; 7: 475-480.
• Kokron C, Errante PR, Barros MT, Baracho GV, Camargo MM, Kalil J et
al. Clinical and laboratory aspects of common variable
immunodeficiency. An Acad Bras Cienc 2004; 76(4): 707-26.
• Lindegren M, Kobrynski L, Rasmussen S, Moore C, Grosse S,
Vanderford M et al. Applying Public Health Strategies to Primary
Immunodeficiency Diseases. A potential approach to Genetic Disorders.
MMWR Recommendations and Reports 2004/53(rr01); 1-29.
• Nowak-Wegrzyn A, Crawford T, Winkelstein J, Carson K, Lederman H.
Immunodeficiency and Infection in Ataxia-Telangiectasia. J Pediatrics
2004; 144: 505-511.
• Ochs H, Thrasher A. The Wiskott-Aldrich syndrome. J Allergy Clin
Immunol 2006; 117:725-738.
• Padeh S, Berkun Y. Auto-inflamatory Fever Syndromes. Rheum Dis Clin
N AM 2007; 33:585-623.
• Pérez N, Liberatore D, Oleastro M, Bedzrodnik L, Rosenzweig S,
Cantisano C et al. Registro Argentino de inmunodeficiencias
primarias.Segundo informe. Arch Argent Pediatr 2007; 105(5):453-460.
• Rodrigues J. Estudo retrospectivo de crianças com suspeita de
imunodeficiência celular ou humoral. Pediatr (São Paulo) 1981; 3:234240.
• Roifman C, Gu Y, Cohen A. Mutations in the RNA component of RNase
mitochondrial RNA processing might cause Omenn syndrome. J Allergy
Clin Immunol 2006; 117: 897-903.
• Rosen FS, Eibl M, Roifman C, Fischer A, Volanakis J, Aiuti F et al.
Primary Immunodeficiency Diseases. Report of an IUIS Scientific
Committee. Clin Exp Immunol 1999; 118 (Supl. 1): 1-28.
• Rosenzweig S, Holland S. Phagocyte Immunodeficiencies and Their
infections. J Allergy Clin Immunol 2004, 113:620-626
• Secretaria de Atenção à Saúde. Portaria no 745 de 22 de dezembro de
2005.http://dtr2001.saude.gov.br/sas/PORTARIAS/Port2005/PT-745.htm
(acessado em 09/01/2007)
• Seger R. Modern Management of Chronic Granulomatous Disease. Br J
Haematol 2008, 140: 255-266
• Shearer WT, Cunningham-Rundles C, Ochs H. Primary
immunodeficiency:Looking backwards, looking forwards. J Allergy Clin
Immunol 2004; 113:607-9.
• Shiroma N, Omi T, Hasegawa H, Nagashima K, Ohta T. A Case of Xlinked Agammaglobulinemia With Progressive Encephalitis. Pediatr
Neurol 2004; 31: 371-3.
• Shulman, Ronca, Bukuvalas. Primary Immune Deficiencies Diseases in
America. The First National Survey of Patients and Specialists. 1995.
http://www.primaryimmune.org Sd.
• Stiehm ER, Ochs HD, Wilkenstein JA. Immunologic Disorders in Infants
& Children 5a ed. Philadelphia, W.B. Saunders, 2004.
• Su H, Lenardo M. Genetic Defects of Apoptosis and Primary
Immunodeficiency. Immunol Allergy Clin N Am 2008; 28: 329-351.
• Verbsky J, Grossman W. Cellular and Genetic bases of Primary Immune
Deficiencies. Pediatric Clin N Am 2006;53:649-684.
• Webster AD. Virus infections in primary immunodeficiency. J Clin Pathol
1994; 47:965-7.
• Weller C, Banker-Fulbright J. Commom Variable Immunodeficiency:
Tests Indications and Interpretations. Mayo Clin Proc 2005; 80(9): 11871200.
• Wen L, Atkinson J, Giclas P. Clinical and laboratory evaluation of
complement deficiency. J Allergy Clin Immunol 2004; 113: 585-593.
• Wood P, Stanworth S, Burton J, Jones A, Peckham D, Green T et al.
Recognition, clinical diagnosis and management of patients with primary
antibody deficiencies: a systematic review. Clin Exp Immunol 2007;
149:410-423
• Yong P, Tarzi M, Chua I, Grimbacher B, Chee R. Commom variable
Immunodeficiency: An Update on Etiology and Management. Immunol
Allergy Clin N Am 2008; 28: 367-386.
•
Zelazco M, Carneiro-Sampaio M, Luigi MC, Olarte DG, Madrigal OP,
Perez RB et al. Primary Immunodeficiency Diseases in Latin America:
First Report from Eight Countries Participating in the LAGID. J Clin
Immunol 1998; 18: 161-6.
CAPÍTULO 11- ANEXOS
ANEXO 1
SINAIS DE ALERTA DA FUNDAÇÃO JEFFREY MODELL
ANEXO 2
FORMULÁRIO DE PRIMEIRA VEZ UTILIZADO NO AMBULATÓRIO DE
ALERGIA E IMUNOLOGIA DO IPPMG
ANEXO 3
CLASSIFICAÇÃO DE GEHA ET AL, 2007.
CAPÍTULO 12 - APÊNDICES
APÊNDICE 1
I.
FERRAMENTA DE COLETA
Instrumento de coleta de dados:
PERFIL CLÍNICO E LABORATORIAL DE CRIANÇAS COM SUSPEITA DE
IMUNODEFICIÊNCIA PRIMÁRIA ATENDIDAS EM UM serviço DE
REFERÊNCIA DO RIO DE JANEIRO
Nome:________________________________________________
Registro:________________
Número:________ Data de nascimento: ___/___/___
GÊNERO (
MASC
Data de admissão no ambulatório de imunodeficiência: ___/___/___
Data dos primeiros sintomas:___/___/___ (ou idade_________)
Origem:
( ) RJ – Capital ( ) RJ – Município
_______________
( ) Outro Estado – Qual? ________
(
) Outro País – Especificar ________________________________
) FEM (
)
Motivo do
encaminhamento:_____________________________________________________
______________________________________________________________________
_______
______________________________________________________________________
_______
Origem do encaminhamento:
( ) interno - IPPMG- especialidade_______________________
( ) ambulatório ( ) Enfermaria
( ) externo (rede SUS) Especificar___________________________________
( ) externo (Hospital Privado)
( ) externo (Consultório Privado)
( )outros
Pais Consangüíneos : ( ) sim ( ) não ( ) desconhecido
História familiar de imunodeficiência, óbitos prematuros ou familiares com
infeções de repetição:
( )sim ( ) não
HPP:
1. Infecções:
Tipo
Pneumonia
Sinusite
Otite Média
Diarréia
Meningite
Sepse
Osteomielite
Outros:
Número de vezes
2. Internações: ( )sim ( )não
Média________
3. Internações em CTI : ( )sim
________
Patógeno
quantas:____________ Duração
( )não
quantas:_______Duração Média
4. Reação à vacinação BCG: ( )sim ( )não qual:__________
5. Necessidade de tratamento específico após reação adversa ao BCG? ( )sim (
)não
6. Cicatriz de BCG: ( ) presente (
registrado
) ausente (
) não verificado/não
HIPÓTESES DIAGNÓSTICAS
A) ________________________________________________________
B) ________________________________________________________
C) ________________________________________________________
CONDUTA DIAGNÓSTICA (INICIAL)
Hemograma
CH50
IgA
C1INH
IgG
G6PD
IgE
ADA
IgM
ANA
Sub-classes de
IgG
FR
DHR
Isohemaglutininas
CD4
Mielograma/BMO
Res. à vacina antiCD8
pneumococo
CD19
Teste de Coombs
CD56
Teste de Linfoproliferação
C3
Teste do Suor
C4
Teste ID
REFLUXO GASTRO-ESOFÁGICO – ( ) sim ( ) não
Sorologia Anti-HIV
Teste de Puntura
Raio-X de Tórax
TC de Tórax
Raio-X de Cavum
( ) possível
CONDUTA TERAPÊUTICA INICIAL
IVIG
ATB profilático
Expectante
Outros*
Qual______________________
* quais:_________________________________
ACOMPANHAMENTO
1. USO DE IVIG:
• Dose Prescrita: _________________________________________
• Intervalo de Administração Prescrito:_______semanas
• Regularidade ( ) sim ( ) não causa___________________________
• Reações adversas ( )sim TIPO________________________ ( ) não
• Infecções após início de IVIG
TIPO
patógenos isolados
Pneumonia
Otite Média
Sinusite
Diarréia
Sepse
Meningite
• Internações ( ) sim ( ) não
Média________
quantas:________ Duração
• Internações em CTI ( ) sim ( ) não
Média________
quantas:________ Duração
2. ATB profilático:
• Regularidade ( ) sim ( ) não causa _____________________________
• Efeito Adverso ( ) sim qual?_____________________ (não)
• Infecções após início de ATB
TIPO
patógenos isolados
Pneumonia
Otite
Sinusite
Diarréia
Sepse
Meningite
• Internações ( ) sim ( ) não
quantas:________ Duração Média________
• Internações em CTI ( ) sim ( ) não
Média________
quantas:________ Duração
3. PRINCIPAIS RESULTADOS DE EXAMES:
Exame
Hemácias
Hemoglobina
Hematócrito
Leucócitos
Neutrófilos
Linfócitos
Plaquetas
IgA
IgG
IgE
IgM
IgG1
IgG2
IgG3
Resultado (Data/Idade)
Exame
Isohemaglotinina
Mielograma/BMO
Resp. à vacina antipneumococo
Teste de Linfoproliferação
Raio-X de Tórax
TC de Tórax
Raio-X de Cavum
NO - Normal
AN - Anormal
NR - Não realizado
NO AN NR
Exame
IgG4
CD4 (abs)
CD4 (%)
CD8 (abs)
CD8 (%)
CD4/CD8
CD19 (abs)
CD19 (%)
CD56 (abs)
CD56 (%)
C3
C4
CH50
C1INH
Resultado (Data/Idade)
Exame
POS NEG NR
G6PD
ADA
ANA
FR
Teste de Coombs
Teste do Suor
Sorologia AntiHIV
Teste ID
Teste de Puntura
POS - Positivo
NEG - Negativo
NR - Não
realizado
4. SEQUELAS:
Pulmonares
Desnutrição
neurológicas
Ósseas
5. Indicação de citocina terapêutica ( ) sim (
) não
qual __________________________
6. Uso de citocina terapêutica ( ) sim ( ) não qual
__________________________
7. Motivo de Impossilibidade/Irregularidade de uso de citocina/fator de
crescimento prescrito :
( ) alto custo (
( ) Outro
8. TMO
• Indicação
)efeito adverso grave (
) indisponível no Brasil
( ) sim ( ) não
• Realizou ( ) sim ( ) não
• Tipo _________________________________
DIAGNÓSTICO FINAL:
DATA DO DIAGNÓSTICO: ____/_____/______
APÊNDICE 2
FORMULÁRIO DE PRIMEIRA VEZ PROPOSTO
DATA:
NOME:
REGISTRO:
ENDEREÇO:
CIDADE:
TELEFONE:
DATA DE NASCIMENTO:
IDADE:
SETOR DE ENCAMINHAMENTO:
conta própria
IPPMG (enfermaria)
IPPMG (amb)
___________________
rede SUS ___________________
consultório privado
hospital privado
__________
MOTIVO DE ENCAMINHAMENTO:
IDADE DO INÍCIO DOS SINTOMAS:
•
• HDA:
INFECÇÕES NO ULTIMO ANO:
TIPO
Pneumonia
( )sim ( )não
NUMERO DE
VEZES
PATÓGENO
ISOLADO
( )sim ( )não
Sinusite
( )sim ( )não
( )sim ( )não
Otite média aguda
( )sim ( )não
( )sim ( )não
Meningite
bacteriana
( )sim ( )não
Meningite viral
(encefalite)
( )sim ( )não
Osteomielite
( )sim ( )não
( )sim ( )não
Artrite séptica
( )sim ( )não
( )sim ( )não
Tuberculose
( )sim ( )não
( )sim ( )não
ITU
( )sim ( )não
( )sim ( )não
( )sim ( )não
( )sim ( )não
TRATAMENTO
Amigdalite
( )sim ( )não
( )sim ( )não
Estomatite
( )sim ( )não
( )sim ( )não
Monilíase
( )sim ( )não
( )sim ( )não
Abscessos
( )sim ( )não
( )sim ( )não
Celulites
( )sim ( )não
( )sim ( )não
Outras infecções
cutâneas
( )sim ( )não
( )sim ( )não
Diarréia viral
( )sim ( )não
( )sim ( )não
Diarréia parasitária
( )sim ( )não
( )sim ( )não
Diarréia bacteriana
( )sim ( )não
( )sim ( )não
Sepse
( )sim ( )não
( )sim ( )não
Outras infecções
virais
( )sim ( )não
( )sim ( )não
Outras infecções
( )sim ( )não
( )sim ( )não
•
•
OUTROS SINTOMAS: (febre, artrites, artralgias, vomitos, sintomas neurológicos, ...)
HPP:
1. Comorbidades(doenças diagnosticadas previamente ou em investigação):
- doença autoimune : ( ) sim ( ) não Qual ______________
- neoplasia: ( )sim ( )não Qual: _______________________
-doenças genéticas: ( ) sim ( )não
Qual:________________________________________
- outras: ( )sim ( )não Quais:
_______________________________________________________
2. Infecções anteriores:
TIPO
Pneumonia
( )sim ( )não
NUMERO DE
VEZES
PATÓGENO
ISOLADO
( )sim ( )não
Sinusite
( )sim ( )não
( )sim ( )não
Otite média aguda
( )sim ( )não
( )sim ( )não
Meningite
bacteriana
( )sim ( )não
( )sim ( )não
Meningite viral
(encefalite)
( )sim ( )não
( )sim ( )não
Osteomielite
( )sim ( )não
( )sim ( )não
Artrite séptica
( )sim ( )não
( )sim ( )não
Tuberculose
( )sim ( )não
( )sim ( )não
ITU
( )sim ( )não
( )sim ( )não
Amigdalite
( )sim ( )não
( )sim ( )não
Monilíase
( )sim ( )não
( )sim ( )não
TRATAMENTO
Abscessos
( )sim ( )não
( )sim ( )não
Celulites
( )sim ( )não
( )sim ( )não
Outras infecções
cutâneas
( )sim ( )não
( )sim ( )não
Diarréia viral
( )sim ( )não
( )sim ( )não
Diarréia parasitária
( )sim ( )não
( )sim ( )não
Diarréia bacteriana
( )sim ( )não
( )sim ( )não
Sepse
( )sim ( )não
( )sim ( )não
Outras infecções
virais
( )sim ( )não
( )sim ( )não
Outras infecções
( )sim ( )não
( )sim ( )não
3. Internações:
( )sim ( )não
quantas:
internação em CTI: ( )sim ( )não
Histórico de internações (tempo de internação, motivo e tratamento):
4. Hemotransfusão: ( )sim ( )não
reação transfusional: ( )sim ( )não
Hemoderivado ____________________________ (
)deleucotizado ( ) outros
5. Alergia respiratória:
( )sim ( )não qual:
Classificação:
6. Dermatite atópica:
) irradiado ( )filtrado (
( )sim ( )não classificação:
•
HG/HP:
G___ P___A___
Gestação ( ) sem intercorrências ( )com intercorrências – quais
___________________________________________________________________
__________
Parto ( ) normal ( )cesáreo /( )a termo ( )pre-termo ( ) pós-termo/ Apgar
__/__
( )sem intercorrências ( )com intercorrências – quais
___________________________
________________________________
________________________________
PESO DE NASCIMENTO:
COMPRIMENTO:
PERÍMETRO CEFÁLICO:
•
HF:
Consanguinidade ( )confirmada ( )não ( )suspeita ( )desconhecido
História sugestiva de imunodeficiência ( )sim ( )não
Outras :
•
HA:
Aleitamento materno exclusivo até 6 meses : ( )sim ( )não
Reação à ptn heteróloga: ( )sim ( )não
Alimentação no momento:
Desnutrição: ( )sim ( )não Grau: _____________________________
•
HD:
Adequado ( )sim ( )não
_________________________________________
_________________________________________
_________________________________________
•
HV:
Cartão em dia: ( )sim ( )não- faltam__________________________________________
Vacinas especiais: ( )não ( )sim- quais_______________________________________
Reações vacinais: ( )não ( )sim – quais______________________________________
EXAME FÍSICO:
PESO:
percentil
CP:
CICATRIZ DE BCG: ( )PRESENTE ( )AUSENTE
ECTOSCOPIA:
PELE E FÂNEROS:
Quantas :
ACV:
AR:
ABDOMEN:
MEMBROS/OSTEOARTICULAR:
URO-GENITAL:
NEUROLÓGICO:
LINFONODOS:
OROFARINGE (dentição, tonsilas, mucosa oral e jugal):
OTOSCOPIA:
EXAMES TRAZIDOS PREVIAMENTE (especificar tipo, data e resultados):
IMPRESSÃO DIAGNÓSTICA:
cid 10CONDUTA (diagnóstica e terapêutica):
RETORNO:
REVISÃO DE SINAIS DE ALERTA:
Duas ou mais pneumonias no último ano
quatro ou mais otites no último ano
estomatites de repetição ou minilíase por mais de 2 meses
abscessos de repetição ou ectima
1 episódio de infecção sistêmica grave
infecções intestinais de repetição/diarréia crônica
asma grave, doença do colágeno oudoença autoimune
efeito adverso ao BCG e/ou infecçao por micobactéria
fenótipo clínico sugestivo de síndrome associada a
imunodeficiência
história familiar de imunodeficiência
2 ou mais sinusites graves no último ano
dois ou mais meses de uso de antibióticos sem efeito
desenvolvimento pondero estatural inadequado
necessidade de antibióticoterapia venosa para cura de infecções
SEGUIMENTO
DATA:
NOME:
HIPÓTESE DIAGNÓSTICA EM INVESTIGAÇÃO: cid 10MEDICAÇÕES EM USO (quais e regularidade do uso):
INTERCORRÊNCIAS (infecções,
tratamentos, internações):
EXAMES REALIZADOS COM RESULTADOS:
EXAME FÍSICO:
PesoCompPercentilDesnutrição: ( )sim ( )não Grau:
IMPRESSÃO DIAGNÓSTICA:
CONDUTA:
RETORNO
REGISTRO:
IDADE: